- Introduction
- Automating Batch-Tag Searching
- Command Line with a mySQL Database
- Command Line without a mySQL Database
- Parameters for Other Protein Prospector Programs
- MS-Fit
- MS-Tag
- MS-Seq
- MS-Pattern
- MS-Homology
- MS-Digest
- MS-Bridge
- MS-NonSpecific
- MS-Product
- MS-Comp
- DB-Stat
- MS-Isotope
- MS-Filter
- FA-Index - Create Indicies for New Database
- FA-Index - Database Summary Report
- FA-Index - Create Subset Database
- FA-Index - Create Subset Database with Indices from Saved Hits
- FA-Index - Create or Append to User Database
Most of the programs in the Protein Prospector package are implemented according to the Common Gateway Interface (CGI) standard. The parameters for the programs are thus a set of name-value pairs. These can either be provided from a HTML form, a script that mimics the CGI format or from the command line.
A detailed description of the CGI format is beyond the scope of this document. Protein Prospector should be able to recognize forms where the method is either GET or POST. If the method is POST then the enctype attribute can either be multipart/form-data or text/plain. A useful book on CGI Programming is:
CGI Programming on the World Wide Web, Shishir Gundavaram, O'Reilly, 1996
The programs can be run from the command line by two different methods either as a long list of name value pairs:
$ programName - name1=value1 name2=value2 ............... nameN=valueN
or with the parameters are stored in an XML file:
$ programName -f params.xml name1=value1 name2=value2 ..... nameN=valueN
For example to index a database using FA-Index on a Windows system:
faindex.cgi - create_database_indicies=1 database=SwissProt.11.02
On a UNIX system you should use a command of the following form (run from the directory):
./faindex.cgi - create_database_indicies=1 database=SwissProt.11.02
Note that the first argument has to be a dash character.
For the case where the parameters are stored in an XML file any parameters submitted on the command line will override those in the file. The format of the xml file should be as follows:
<?xml version="1.0" encoding="UTF-8"?> <parameters> <name1>value1</name1> <name2>value2</name2> ..... <nameN>valueN</nameN> </parameters>
The second version may be preferred if there are a lot of parameters or the environment you submit the command from imposes a maximum length for a command. A second alternative in this case would be to use a Perl script:
$c = "programName"; $c .= " - "; $c .= "name1=value1"; $c .= " "; $c .= "name2=value2"; $c .= " "; ........ $c .= "nameN=valueN"; $c .= " "; system "$c";
Certain characters need to be escaped when included in a value. These are uppercase hexadecimal ASCII values preceded by a percent sign. If the parameters are stored in an XML file they don't strictly need to be escaped like this but can be. Some commonly used escape codes are listed in the table below:
| space | %20 |
| line end | %0D%0A |
| comma | %2C |
Some parameters such as would appear as multiple choice menus or tick boxes on a HTML form may be specified multiple times with different values. Eg in an XML parameter file context:
..... <mod_AA>Peptide%20N-terminal%20Gln%20to%20pyroGlu</mod_AA> <mod_AA>Oxidation%20of%20M</mod_AA> <mod_AA>Protein%20N-terminus%20Acetylated</mod_AA> .....
or in a command line context:
$ command.cgi - it=a it=b it=y ...................
If the parameter is entered via a text box on a HTML form then its value can extend over several lines.
In an XML parameter file this could be specified thus:
<accession_nums>P40069 P15180 P11484</accession_nums>
Or thus:
<accession_nums>P40069%0D%0AP15180%0D%0AP11484</accession_nums>
In a list of command line arguments it would be specified as follows:
$ command.cgi - accession_nums=P40069%0D%0AP15180%0D%0AP11484 .....
To make a project you need to use the msform.cgi binary. The parameters used are shown below.
| name | Default Value | Valid Values |
|---|---|---|
| data_file_list | None Defined | Paths to centroid data files in the centroid data repository. |
| project_name | "" | text |
| form | "" | needs to be set to makeproject |
| instrument_name | "" | valid text strings from params/instrument.txt |
| user | "" | valid user from the database |
| write_file | false | needs to be set to true |
For example you could put the following into a file called test.xml:
<?xml version="1.0" encoding="UTF-8"?> <parameters> <data_file_list>Q-STAR1/2008/06/F3.mgf</data_file_list> <data_file_list>Q-STAR1/2008/06/F4.mgf</data_file_list> <project_name>test</project_name> <form>makeproject</form> <instrument_name>ESI-Q-TOF</instrument_name> <user>pp</user> <write_file>1</write_file> </parameters>
and run the command as follows:
$ msform.cgi -f test.xml
To run a Batch-Tag search you need to use the msbatch.cgi binary. The parameters used are shown below.
| name | Default Value | Valid Values |
|---|---|---|
| search_name | "" | needs to be set to batchtag |
| report_title | "" | text |
| database | Non selected | valid prefixes: Genpept, gen, SwissProt, swp, Owl, owl, UniProt, Ludwignr, NCBInr, nr, dbEST, dbest, pdbEST, pdbest, IPI, ipi, DA, DN, PA, PN, pDA, pDN, Pdefault, Ddefault, pDdefault. User Protein is another possible selection. Multiple databases may be specified. |
| user_protein_sequence | "" | protein as a text string |
| dna_frame_translation | 3 | 6, 3, -3, 1, -1 |
| species | All | valid text strings from params/taxonomy.txt, params/taxonomy_groups.txt or All |
| output_filename | "" | text |
| enzyme | Trypsin | valid text strings from params/enzyme.txt or params/enzyme_comb.txt |
| allow_non_specific | at 0 termini | at 0 termini, at 1 termini, at 2 termini, at N termini, at C termini, N termini-1=D |
| missed_cleavages | 1 | integer |
| const_mod | None defined | valid text strings formed from the information in params/usermod.txt. |
| msms_prot_low_mass | 1000 | integer |
| msms_prot_high_mass | 100000 | integer |
| msms_full_mw_range | 0 | 0, 1 |
| low_pi | 3.0 | double |
| high_pi | 10.0 | double |
| full_pi_range | 0 | 0, 1 |
| results_from_file | 0 | 0, 1 |
| results_input_dir | results_" + value of input_program_name parameter eg: results_msfit | directory name |
| input_program_name | msfit | msfit, mstag, mspattern, mshomology, msseq |
| input_filename | "" | text |
| species_names | None Defined | list of text strings |
| species_remove | 0 | 0, 1 |
| names | None Defined | list of text strings |
| accession_nums | None Defined | list of text strings |
| add_accession_numbers | None Defined | list of text strings |
| bond_score | 0 | 0,1 |
| expect_calc_method | None | None, Linear Tail Fit, Linear Tail Fit With Consistent Seeds |
| msms_precursor_charge_range | "" | List of charges required separated by spaces |
| parent_mass_convert | monoisotopic | monoisotopic average Par(mi)Frag(av) Par(av)Frag(mi) |
| msms_parent_mass_tolerance | 0.5 | double |
| msms_parent_mass_tolerance_units | Da | Da, %, ppm, mmu |
| msms_parent_mass_systematic_error | 0.0 | double |
| fragment_masses_tolerance | 0.5 | double |
| fragment_masses_tolerance_units | Da | Da, %, ppm, mmu |
| msms_mod_AA | None defined | valid text strings formed from the information in params/usermod.txt. |
| msms_max_modifications | 1 | integer |
| msms_max_peptide_permutations | "" | Left blank or non zero positive integer |
| mod_start_nominal | 0 | integer |
| mod_end_nominal | 0 | integer |
| mod_defect | 0.0 | double |
| mod_comp_ion | None defined | A,C,D,E,F,G,H,I,K,L,M,N,P,Q,R,S,T,V,W,Y |
| mod_n_term | 0 | 0, 1 |
| mod_c_term | 0 | 0, 1 |
| mod_neutral_loss | 0 | 0, 1 |
| mod_uncleaved | 0 | 0, 1 |
| msms_pk_filter | Max MSMS Pks | Max MSMS Pks or Max MSMS Pks / 100 Da |
| msms_max_peaks | "" | integer |
| msms_search_type | None defined | valid text strings formed from the file params/homology.txt. |
| data_source | Data Paste Area | need to be set to "List of Files". |
| instrument_name | "" | valid text strings from params/instrument.txt |
| project_name | "" | text |
| user | "" | valid user from the database |
| comment | "" | text |
| max_hits | 200 | integer |
| msms_max_reported_hits | 50 | integer |
| script_filename | "" | text |
| use_instrument_ion_types | 0 | 0 or 1 |
| save_params | 0 | 0 or 1 |
For example you could put the following into a file called test.xml:
<?xml version="1.0" encoding="UTF-8"?> <parameters> <allow_non_specific>at 0 termini</allow_non_specific> <const_mod>Carbamidomethyl (C)</const_mod> <data_source>List of Files</data_source> <database>SwissProt.2008.08.20</database> <dna_frame_translation>3</dna_frame_translation> <enzyme>Trypsin</enzyme> <expect_calc_method>Linear Tail Fit</expect_calc_method> <fragment_masses_tolerance>300</fragment_masses_tolerance> <fragment_masses_tolerance_units>ppm</fragment_masses_tolerance_units> <full_pi_range>1</full_pi_range> <high_pi>10.0</high_pi> <instrument_name>ESI-Q-TOF</instrument_name> <low_pi>3.0</low_pi> <max_hits>9999999</max_hits> <missed_cleavages>1</missed_cleavages> <msms_full_mw_range>1</msms_full_mw_range> <msms_max_modifications>2</msms_max_modifications> <msms_max_reported_hits>5</msms_max_reported_hits> <msms_mod_AA>Acetyl (Protein N-term)</msms_mod_AA> <msms_mod_AA>Gln->pyro-Glu (N-term Q)</msms_mod_AA> <msms_mod_AA>Met-loss (Protein N-term M)</msms_mod_AA> <msms_mod_AA>Oxidation (M)</msms_mod_AA> <msms_parent_mass_systematic_error>0</msms_parent_mass_systematic_error> <msms_parent_mass_tolerance>200</msms_parent_mass_tolerance> <msms_parent_mass_tolerance_units>ppm</msms_parent_mass_tolerance_units> <msms_precursor_charge_range>2 3</msms_precursor_charge_range> <msms_prot_high_mass>125000</msms_prot_high_mass> <msms_prot_low_mass>1000</msms_prot_low_mass> <output_filename>results</output_filename> <parent_mass_convert>monoisotopic</parent_mass_convert> <project_name>test</project_name> <report_title>BatchTag</report_title> <search_name>batchtag</search_name> <species>SACCHAROMYCES CEREVISIAE</species> <use_instrument_ion_types>1</use_instrument_ion_types> <user>pbaker</user> </parameters>
and run the command as follows:
$ msbatch.cgi -f test.xml
To run Search Compare you need to use the searchCompare.cgi binary. The parameters used are shown below.
| name | Default Value | Valid Values |
|---|---|---|
| save_format | HTML | HTML, Tab delimited text, Filtered Peak Lists, MS-Viewer Files, pepXML Filtered, pepXML Unfiltered |
| accession_nums | None Defined | list of text strings |
| remove | 0 | 0 or 1 |
| multi_sample | 0 | 0 or 1 |
| id_filter_list | None Defined | List of integers |
| preferred_species | None Defined | List of text strings |
| min_best_disc_score_ESI_Q_TOF | 0.0 | double |
| min_best_disc_score_ESI-ION-TRAP-low-res | 0.0 | double |
| min_best_disc_score_ESI-FT-ICR-CID | 0.0 | double |
| min_best_disc_score_ESI-FT-ICR-ECD | 0.0 | double |
| min_best_disc_score_MALDI-Q-TOF | 0.0 | double |
| min_best_disc_score_MALDI-TOFTOF | 0.0 | double |
| min_pp_protein_score | 0.0 | double |
| min_pp_peptide_score | 0.0 | double |
| max_protein_evalue | 0.01 | double |
| max_peptide_evalue | 0.01 | double |
| keep_replicates | 0 | 0 or 1 |
| best_disc_only | 0 | 0 or 1 |
| disc_score_graph | 0 | 0 or 1 |
| comp_mask_type | AND | OR, AND |
| comp_ion | None defined | A,C,D,E,F,G,H,I,K,L,M,N,P,Q,R,S,T,V,W,Y and valid text strings formed from the information in params/usermod.txt. |
| mass_comp_list | None defined | List of doubles |
| report_m_plus_h | 0 | 0 or 1 |
| report_m_over_z | 0 | 0 or 1 |
| report_charge | 0 | 0 or 1 |
| report_m_plus_h_calc | 0 | 0 or 1 |
| report_m_over_z_calc | 0 | 0 or 1 |
| report_intensity | 0 | 0 or 1 |
| report_error | 0 | 0 or 1 |
| report_unmatched | 0 | 0 or 1 |
| report_num_pks | 0 | 0 or 1 |
| report_rank | 0 | 0 or 1 |
| report_score | 0 | 0 or 1 |
| report_score_diff | 0 | 0 or 1 |
| report_expectation | 0 | 0 or 1 |
| report_p_value | 0 | 0 or 1 |
| report_nlog_p_value | 0 | 0 or 1 |
| report_num_precursor | 0 | 0 or 1 |
| report_gradient | 0 | 0 or 1 |
| report_offset | 0 | 0 or 1 |
| report_disc_score | 0 | 0 or 1 |
| report_repeats | 0 | 0 or 1 |
| report_prot_score | 0 | 0 or 1 |
| report_num_unique | 0 | 0 or 1 |
| report_peptide_count | 0 | 0 or 1 |
| report_best_score | 0 | 0 or 1 |
| report_best_expect | 0 | 0 or 1 |
| report_coverage | 0 | 0 or 1 |
| report_best_disc_score | 0 | 0 or 1 |
| report_peptide | 0 | 0 or 1 |
| peptide_mod_type | Variable Mods Only | Off, Mods In Peptide, Variable Mods Only, Constant Mods Only, All Mods (1 column), All Mods (2 columns) |
| report_var_mod | 0 | 0 or 1 |
| report_protein_mod | 0 | 0 or 1 |
| slip_threshold | 0.0 | double |
| report_length | 0 | 0 or 1 |
| report_composition | 0 | 0 or 1 |
| report_start_aa | 0 | 0 or 1 |
| report_end_aa | 0 | 0 or 1 |
| report_previous_aa | 0 | integer |
| report_next_aa | 0 | integer |
| report_missed_cleavages | 0 | 0 or 1 |
| report_mass_mod | 0 | 0 or 1 |
| report_time | 0 | 0 or 1 |
| report_number | 0 | 0 or 1 |
| report_accession | 0 | 0 or 1 |
| report_uniprot_id | 0 | 0 or 1 |
| report_gene_name | 0 | 0 or 1 |
| report_prot_len | 0 | 0 or 1 |
| report_mw | 0 | 0 or 1 |
| report_pi | 0 | 0 or 1 |
| report_species | 0 | 0 or 1 |
| report_name | 0 | 0 or 1 |
| report_links | 0 | 0 or 1 |
| raw_type | "" | MS Precursor, MS Full Scan, MS/MS, Quantitation |
| quan_type | "" | valid text strings formed from the information in params/quan.xml and params/quan_msms.xml. |
| rep_q_median | 0 | 0 or 1 |
| rep_q_iqr | 0 | 0 or 1 |
| rep_q_mean | 0 | 0 or 1 |
| rep_q_n_sdv | 2.0 | double |
| rep_q_stdev | 0 | 0 or 1 |
| rep_q_num | 0 | 0 or 1 |
| rep_intensity | 0 | 0 or 1 |
| intensity_threshold | 0.0 | double |
| rep_resolution | 0 | 0 or 1 |
| rep_a_lh_int | 0 | 0 or 1 |
| rep_area | 0 | 0 or 1 |
| area_threshold | 0.0 | double |
| rep_a_lh_area | 0 | 0 or 1 |
| rep_snr | 0 | 0 or 1 |
| snr_threshold | 0.0 | double |
| rep_n_mean | 0 | 0 or 1 |
| rep_n_stdev | 0 | 0 or 1 |
| rt_int_start | 0.0 | double |
| rt_int_end | 0.0 | double |
| resolution | 10000.0 | double |
| percent_C13 | 100.0 | double |
| percent_N15 | 100.0 | double |
| percent_O18 | 100.0 | double |
| purity_correction | 0 | From formulae, Default, No correction and options from purity files. |
| database_type | None defined | valid prefixes: Genpept, gen, SwissProt, swp, Owl, owl, UniProt, Ludwignr, NCBInr, nr, dbEST, dbest, pdbEST, pdbest, IPI, ipi, DA, DN, PA, PN, pDA, pDN, Pdefault, Ddefault, pDdefault. |
| report_type | Protein | Protein, Peptide, Time, False Positive Rate |
| report_homologous_proteins | Interesting | All, Interesting, None |
| report_hits_type | "" | Union, Intersection, Difference |
| sort_type | "" | Discriminant Score, Peptide Score, Expectation Value, Start Residue, Fraction/RT, RT, m/z, M+H, Intensity, Error, Time, Charge/M+H, Mass Mod |
| sort_type2 | "" | "", Discriminant Score, Peptide Score, Expectation Value, Start Residue, Fraction/RT, RT, m/z, M+H, Intensity, Error, Time, Charge/M+H, Mass Mod |
| msms_pk_filter | Max MSMS Pks | Max MSMS Pks, Max MSMS Pks / 100 Da or Unprocessed MSMS |
| unmatched_spectra | 0 | 0 or 1 |
| msms_max_peaks | "" | integer |
| msms_max_reported_hits | 50 | integer |
| data | None defined | List of search job keys from mySQL database for searches that you want to compare. The relevant database field is search_jobs.search_job_key |
| parent_mass_convert | monoisotopic | monoisotopic average Par(mi)Frag(av) Par(av)Frag(mi) |
| run_msproduct | 0 | 0 or 1 |
| save_params | 0 | 0 or 1 |
| output_directory | "" | directory |
| output_filename | "" | filename for Tab Delimited Text output or zip archive name for pepXML output |
For example you could put the following into a file called test.xml:
<?xml version="1.0" encoding="UTF-8"?> <parameters> <save_format>Tab delimited text</save_format> <min_best_disc_score_ESI_Q_TOF>0.0</min_best_disc_score_ESI_Q_TOF> <min_pp_protein_score>22.0</min_pp_protein_score> <min_pp_peptide_score>15.0</min_pp_peptide_score> <max_protein_evalue>0.01</max_protein_evalue> <max_peptide_evalue>0.01</max_peptide_evalue> <keep_replicates>0</keep_replicates> <best_disc_only>1</best_disc_only> <disc_score_graph>0</disc_score_graph> <report_m_plus_h>0</report_m_plus_h> <report_m_over_z>1</report_m_over_z> <report_charge>1</report_charge> <report_m_plus_h_calc>0</report_m_plus_h_calc> <report_m_over_z_calc>0</report_m_over_z_calc> <report_intensity>0</report_intensity> <report_error>1</report_error> <report_unmatched>0</report_unmatched> <report_num_pks>0</report_num_pks> <report_rank>0</report_rank> <report_score>1</report_score> <report_score_diff>0</report_score_diff> <report_expectation>1</report_expectation> <report_num_precursor>0</report_num_precursor> <report_disc_score>0</report_disc_score> <report_repeats>1</report_repeats> <report_prot_score>0</report_prot_score> <report_num_unique>1</report_num_unique> <report_peptide_count>0</report_peptide_count> <report_best_score>0</report_best_score> <report_best_expect>1</report_best_expect> <report_coverage>1</report_coverage> <report_best_disc_score>1</report_best_disc_score> <report_peptide>1</report_peptide> <peptide_mod_type>Variable Mods Only</peptide_mod_type> <report_protein_mod>1</report_protein_mod> <slip_threshold>6</slip_threshold> <report_length>0</report_length> <report_composition>0</report_composition> <report_start_aa>0</report_start_aa> <report_end_aa>0</report_end_aa> <report_previous_aa>1</report_previous_aa> <report_next_aa>1</report_next_aa> <report_missed_cleavages>0</report_missed_cleavages> <report_mass_mod>0</report_mass_mod> <report_time>1</report_time> <report_number>1</report_number> <report_accession>1</report_accession> <report_gene_name>0</report_gene_name> <report_prot_len>0</report_prot_len> <report_mw>1</report_mw> <report_pi>0</report_pi> <report_species>1</report_species> <report_name>1</report_name> <report_links>1</report_links> <database_type>SwissProt</database_type> <report_type>Peptide</report_type> <report_homologous_proteins>Interesting</report_homologous_proteins> <report_hits_type>Union</report_hits_type> <sort_type>Expectation Value</sort_type> <sort_type_2></sort_type_2> <data>VzTrrrtteaD7HySm</data> <parent_mass_convert>monoisotopic</parent_mass_convert> </parameters>
and run the command as follows:
$ searchCompare.cgi -f test.xml > temp.txt
The jobStatus.cgi binary can be used to control or output job status. The parameters for the program are shown below:
| name | Default Value | Valid Values |
|---|---|---|
| abort | 0 | 0 or 1 |
| search_key | "" | search keys stored in the mySQL database |
For example to output the job status of a given job enter the following command:
$ jobStatus.cgi - search_key=VzTrrrtteaD7HySm
To abort a given job:
$ jobStatus.cgi - search_key=VzTrrrtteaD7HySm abort=1
The msform.cgi binary can be used to delete projects and results and to import or export projects.
| name | Default Value | Valid Values |
|---|---|---|
| form | "" | needs to be set to results_management |
| action | Delete | Delete, Import, Export, Compress, Uncompress or Check |
| user | "" | valid user from the database |
| project_name | None defined | List of projects to delete |
| results_file | None defined | List of results files to delete. These need to be specified in the form results/project |
| data_export | All Data | All Data, Peaklists Only or No Data |
| new_project_name | "" | text |
| upload_temp | "" | file name |
| date_filter | All Projects | All Projects, Projects Created Between or Projects Accessed Between |
| start_month | current month | 01, 02, 03, 04, 05, 06, 07, 08, 09, 10, 11, 12 |
| end_month | current month | 01, 02, 03, 04, 05, 06, 07, 08, 09, 10, 11, 12 |
| start_year | current year | list of years from 2007 |
| end_year | current year | list of years from 2007 |
| uploads_optional | 1 | needs to be set to 1 |
For example to delete some of the pp's projects from the database put the following commands into a file called test.xml:
<?xml version="1.0" encoding="UTF-8"?> <parameters> <action>Delete</action> <project_name>test</project_name> <project_name>test2</project_name> <form>results_management</form> <user>pp</user> </parameters>
and run the command as follows:
$ msform.cgi -f test.xml
If you delete a project then all the results for that project are also deleted.
If you just want to delete a set of results then the parameter file might look as follows:
<?xml version="1.0" encoding="UTF-8"?> <parameters> <action>Delete</action> <results_file>test/results1</results_file> <form>results_management</form> <user>pp</user> </parameters>
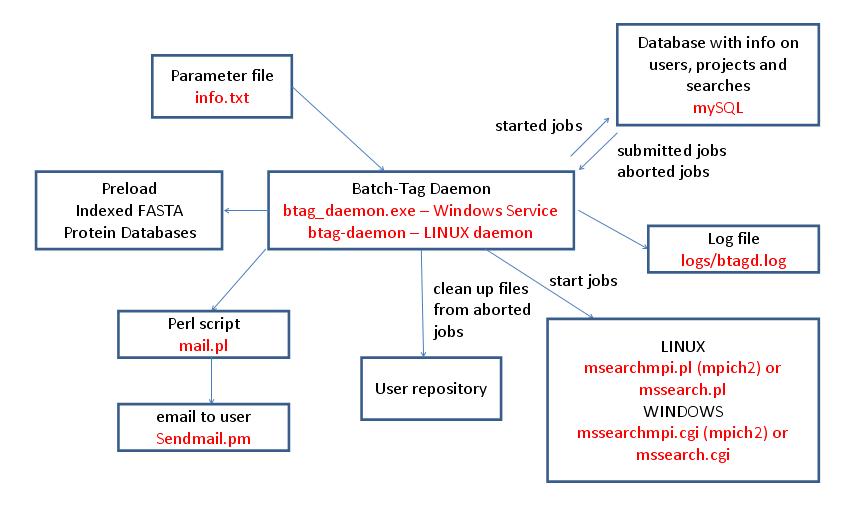
The Batch-Tag Daemon is the program that controls the running of Batch-Tag searches in a Protein Prospector system. It runs as a service on a Windows computer (btag_daemon.exe) or as a daemon on a LINUX computer (btag-daemon). Multiple computers can run Batch-Tag Daemons at the same time. Its setup parameters are contained in the file info.txt which it reads on startup or on receipt of a HUP signal (LINUX only). It periodically checks a mySQL database for newly submitted or aborted searches. It will start a search if there are sufficient resources; otherwise the searches are queued or run by another Batch-Tag Daemon. On LINUX systems searches are run using a Perl script (mssearchmpi.pl or mssearch.pl) whereas on Windows systems the executable program is called directly (mssearchmpi.cgi or mssearch.cgi). Whether mssearchmpi or mssearch is used depends on whether the system is configured to run searches using multiple processes via MPICH2. If searches are aborted or finish unexpectedly the Batch-Tag Daemon is responsible for cleaning up the files in the user repository. It is possible to configure the daemon to send email to users when searches have finished via the Perl script mail.pl. It is also capable of preloading indexed protein databases into memory. It logs its activity to the file logs/btagd.log.
Peak Spotter is used to extract data from an ABI 4700/4800 TOF-TOF Oracle database (version 3). It doesn't require the rest of the Protein Prospector installation to work. However the Oracle client software needs to be installed to allow the database to be accessed. The program can work on UNIX type platforms as well as Windows ones. The program parameters are as follows:
| Parameter | Default Value | Valid Values |
|---|---|---|
| server_name | "" | text |
| username | TSQUARED | text |
| password | TS | text |
| spot_set_list | 0 | 0 or 1 |
| run_number | 0 | integer |
| all_runs | 0 | 0 or 1 |
| write_raw_ms | 0 | 0 or 1 |
| write_raw_msms | 0 | 0 or 1 |
| retain_isotopes | 0 | 0 or 1 |
| minimum_area | 100.0 | floating point number |
| intensity_type | Height | Height, Area, Cluster Area, Signal to Noise |
| spot_set_names | None defined | Spot sets names from the database |
| centroid_dir | "" | valid directory name |
| centroid_filename | output.txt | valid filename |
| raw_dir | "" | valid directory name |
If the parameter is set to the default value then it does not need to be specified.
server_name
This is the server name as defined in the Oracle client file tnsname.org. This parameter is always required.
username
A username to log in to the database. This parameter is always required.
password
A password for the given username. This parameter is always required.
spot_set_list
This parameter is only required if you want to get a list of spot sets in the database. If you set this parameter to 1 then all the parameters below should not be used.
run_number
The run number can be specified if you just want to extract the data for a given run.
all_runs
This parameter should be set unless you just want to extract the data for a single run.
write_raw_ms
If you set this parameter the raw MS data is also extracted from the database. A separate T2D file is extracted for each spectrum. You need to extract this if you want to do quantitation, such as SILAC, that uses the MS data. You should also extract it if you want to include columns in the Search Compare report, such as intensity, which are calculated from the raw data or to be able to see the parent ion raw data by clicking on the m/z column in the Search Compare peptide report. These files are not used by the database search program.
write_raw_msms
If you set this parameter the raw MSMS data is also extracted from the database. A separate T2D file is extracted for each spectrum. You need to extract this if you want to do quantitation, such as iTRAQ, that uses the MSMS data. You should also extract it if you want to be able to look at the raw data from the Search Compare report. These files are not used by the database search program.
retain_isotopes
If you set this parameter then the isotope peaks are retained in the centroid file. Note that the default setting is for Prospector to allow the ABI software to do the deisotoping. If you want Protein Prospector to do the deisotoping then the Prospector instrument file will need to be set up accordingly.
minimum_area
The minimum area for a peak before it is included in the centroid file. Sometimes the peak lists can be very large if this value isn't set appropriately.
intensity_type
The units for the intensities stored in the centroid file. The possible values for the intensity type are:
- Intensity - The peak intensity of the monoisotopic peak.
- Area - The peak area of the monoisotopic peak.
- Cluster Area - The summed areas of all the peaks in the isotope cluster.
- Signal to Noise - The signal to noise ratio of the monoisotopic peak.
spot_set_names
A list of the names of the spot sets (one per line) to extract from the database. Generally only a single spot set is extracted at one time.
centroid_dir
The directory where you want the centroid file to be created. This directory should exist before running the program. A full path should be given for the directory.
centroid_filename
The name for the centroid file created.
raw_dir
The directory where you want the raw files to be created. This directory should exist before running the program. A full path should be given for the directory.
Examples
To get a list of spot sets from the database:
$ peakSpotter.cgi - server_name=server username=n password=p spot_set_list=1
Alternatively the following could be stored in a file called p.xml:
<?xml version="1.0" encoding="UTF-8"?> <parameters> <server_name>server</server_name> <username>n</username > <password>p</password> <spot_set_list>1</spot_set_list> </parameters>
and the command run as follows:
$ peakSpotter.cgi -f p.xml
To extract a spot set from the database into a Protein Prospector data repository:
Create a file (say p.xml) containing the following:
<?xml version="1.0" encoding="UTF-8"?> <parameters> <server_name>server</server_name> <username>n</username> <password>p</password> <all_runs>1</all_runs> <write_raw_ms>1</write_raw_ms> <write_raw_msms>1</write_raw_msms> <retain_isotopes>0</retain_isotopes> <minimum_area>100.0</minimum_area> <intensity_type>Height</intensity_type> <spot_set_names>User Project 1\test</spot_set_names> <centroid_dir>R:\peaklists\TOFTOF1\2005\11</centroid_dir> <centroid_filename>User Project 1$test.txt</centroid_filename> <raw_dir>R:\raw\TOFTOF1\2005\11\User Project 1$test</raw_dir> </parameters>
and run the command as follows:
$ peakSpotter.cgi -f p.xml
Note that
- The raw directory needs to exist before running peakSpotter.
- Once several spot sets have been extracted you can combine them together to make a Protein Prospector project using the Make Project program. However for this to work the directory where the raw files are going to be stored needs to have the same name as the centroid file (without the .txt suffix). The name doesn't have to correspond to the name of the spot set in the database. The .txt suffix for the centroid file is also currently mandatory.
- The \ character in the original spot set name has been replaced by a $ character so it isn't mistaken for part of the raw directory path.
- Peaklists for both the MS and MSMS spectra are stored in the one centroid file.
- If you extract the raw data then the program will typically take a lot longer to run.
The program wiffToCentroid.exe is a simple command line program to extract mgf (Mascot Generic Format) peak lists from ABI Sciex wiff files. It is compatible with Analyst QS 2.0 and Mascot.dll version 1.1446.0.20. An example use of the program is:
$ wiffToCentroid.cgi C:\wiffs\example.wiff C:\mgfs\example.mgf
The full paths to the wiff file and the mgf file must be specified.
The Analyst service must be running on the computer on which wiffToCentroid is running. The Analyst centroiding parameters stored in the registry are used to do the centroiding.
This program can only run on a Windows computer and does not support a CGI or XML parameter file interface.
In the example given below it is assumed you are working on LINUX in your home directory. The Prospector files are in a subdirectory called Prospector. The Prospector directory has further subdirectories:
peaklists projects reports seqdb temp web xml
Note that if you are intending to run a large number of searches then it is probably best to use more subdirectory levels or repositories as there can be problems if a single directory contains too many files. A data repository for peak list files is described in the Server Administration manual.
If you are running on multiple nodes the table below shows which of these directories needs to be visible to all nodes and which to only the base node. It also shows which directories can be mirrored on the different nodes to potentially reduce the search time.
| Directory | Node Visibility | Mirroring Possible |
|---|---|---|
| peaklists | All | Yes |
| projects | All | Yes |
| reports | N/A | N/A |
| seqdb | All | Yes |
| temp | All | No |
| web | All | Yes |
| xml | Base | N/A |
This sub-directory holds the peak list files. The peak lists for each project are held in separate subdirectories. The peak lists for the example data set is held in the subdirectory test.
The projects sub-directory holds the project files and any associated expectation value files (created during searches).
The reports subdirectory holds the output files from the Search Compare program.
This subdirectory holds the FASTA sequence databases. Any databases used need to be indexed using the FA-Index program before running any Batch-Tag searches.
The temp subdirectory holds the Batch-Tag output files. These are XML files which need to be further processed by the Search Compare program to obtain your final results.
This subdirectory holds the Protein Prospector binaries and parameter files. Any programs need to be run from the cgi-bin subdirectory. The parameter files, which you may need to edit from time to time, are in the params subdirectory.
This subdirectory holds the Batch-Tag and Search Compare search parameter files. Two files bt_test.xml and sc_test.xml are present in the directory as examples.
A project gathers together the peaklists for a particular data set. You can run multiple searches on a given project. When making projects two cases are catered for. Firstly the simple case when all the peak lists are in a single directory. No other files may be present in the directory. Secondly there's the case when the files are located across multiple directories. This might be the case if you have your data in a repository as outlined in the server administration manual. In the examples shown below there are cases where the parameters are specified on the command line and cases where a parameter file is used. Both methods of running a program can be used but the parameter file method is generally preferable if there are a lot of parameters.
./msform.cgi - form=makeproject write_file=1 project_name=test project_filepath=/home/peter/Prospector/projects peak_list_filepath=/home/peter/Prospector/peaklists/test
In the example below the peak list files are actually in the same directory. However this isn't necessary.
If there are only a few peak list files then the paths can be specified on the command line.
./msform.cgi - form=makeproject write_file=1 project_name=test2 project_filepath=/home/peter/Prospector/projects data_file_list=/home/peter/Prospector/peaklists/test/F20130415-02_ITMSms2cid.txt data_file_list=/home/peter/Prospector/peaklists/test/F20130415-03_ITMSms2cid.txt
If there are a lot of peak list files in your data set it is probably more convenient to use a parameter file.
<?xml version="1.0" encoding="UTF-8"?> <parameters> <form>makeproject</form> <write_file>1</write_file> <project_name>test2</project_name> <project_filepath>/home/peter/Prospector/projects</project_filepath> <data_file_list>/home/peter/Prospector/peaklists/test/F20130415-02_ITMSms2cid.txt</data_file_list> <data_file_list>/home/peter/Prospector/peaklists/test/F20130415-03_ITMSms2cid.txt</data_file_list> <data_file_list>/home/peter/Prospector/peaklists/test/F20130415-04_ITMSms2cid.txt</data_file_list> <data_file_list>/home/peter/Prospector/peaklists/test/F20130415-05_ITMSms2cid.txt</data_file_list> <data_file_list>/home/peter/Prospector/peaklists/test/F20130415-06_ITMSms2cid.txt</data_file_list> <data_file_list>/home/peter/Prospector/peaklists/test/F20130415-07_ITMSms2cid.txt</data_file_list> <data_file_list>/home/peter/Prospector/peaklists/test/F20130415-08_ITMSms2cid.txt</data_file_list> <data_file_list>/home/peter/Prospector/peaklists/test/F20130415-09_ITMSms2cid.txt</data_file_list> <data_file_list>/home/peter/Prospector/peaklists/test/F20130415-10_ITMSms2cid.txt</data_file_list> <data_file_list>/home/peter/Prospector/peaklists/test/F20130415-11_ITMSms2cid.txt</data_file_list> <data_file_list>/home/peter/Prospector/peaklists/test/F20130415-12_ITMSms2cid.txt</data_file_list> <data_file_list>/home/peter/Prospector/peaklists/test/F20130415-13_ITMSms2cid.txt</data_file_list> <data_file_list>/home/peter/Prospector/peaklists/test/F20130415-14_ITMSms2cid.txt</data_file_list> <data_file_list>/home/peter/Prospector/peaklists/test/F20130415-15_ITMSms2cid.txt</data_file_list> </parameters>
If the parameter file is called params.xml then the command from the web/cgi-bin directory is:
./msform.cgi -f params.xml
The first step in running a Batch-Tag search is to create an xml file containing the search parameters. An example file called bt_test.xml is supplied in the xml subdirectory.
To run a single processor Batch-Tag search using the example parameter file enter the following command from the web/cgi-bin directory:
./mssearch.cgi -f ../../xml/bt_test.xml
Protein Prospector supports both mpich2 and openmpi. If possible we recommend using openmpi on all new installations. The Protein Prospector binaries need to be compiled with the required MPI option.
If you are running mpich2 then you need to start the MPI daemon before running searches. Also required is a file in your home directory called .mpd.conf. This should set a value for the MPD_SECRETWORD variable. For example it could contain a single line such as:
MPD_SECRETWORD=asecretword
You can start the mpich2 daemon with the following command:
mpd&
The command for stopping the mpich2 daemon is:
mpdallexit
openmpi does not require starting a daemon or a .mpf.conf file.
A search using 9 processors can be run using the following command from the web/cgi-bin directory. Note that the 9 processes includes a root process which doesn't use many resources. Thus you should typically add a processor to account for this.
mpiexec -n 9 ./mssearchmpi.cgi -f ../../xml/bt_test.xml
Note that it may be the case that both mpich2 and openmpi are installed on your server. If this is the case you need to make sure that the correct mpiexec is run. As an example on debian the mpiexec for openmpi is installed in the /usr/bin directory whereas for mpich2 it is installed in /usr/local/bin. One way to thus ensure that the mpiexec in /usr/bin took priority would be to add the following line to your .bashrc file:
PATH="/usr/bin:${PATH}"
You need to login again for this change comes into effect.
The /etc/hosts file is important when using mpi on multiple nodes. It should be defined as follows:
127.0.0.1 localhost 192.168.133.100 ub0 192.168.133.101 ub1 192.168.133.102 ub2 192.168.133.103 ub3
Not like this:
127.0.0.1 localhost 127.0.1.1 ub0 192.168.133.100 ub0 192.168.133.101 ub1 192.168.133.102 ub2 192.168.133.103 ub3
or like this:
127.0.0.1 localhost 127.0.1.1 ub0 192.168.133.101 ub1 192.168.133.102 ub2 192.168.133.103 ub3
As an example the rodin04/rodin05 cluster has the following entries in /etc/hosts.
127.0.0.1 localhost 169.230.19.39 rodin04.ucsf.edu rodin04 169.230.19.40 rodin05.ucsf.edu rodin05
For mpich2 the daemon needs to be started across multiple nodes. In your home directory you need a file called mpd.hosts which lists the hosts. Eg for the UCSF rodin04/rodin05 cluster the file would contain the following lines:
rodin04 rodin05
The mpich2 command for starting the daemon across multiple nodes, which should be run from your home directory, is:
mpdboot --totalnum=2 --verbose
The mdpboot command may ask for passwords for logging into the remote nodes. Note that totalnum argument is used to specify how many lines out of mpd.hosts to process. Thus if you specified this as 1 then the daemon would only start on rodin04.
The mpich2 command to stop the daemon across all nodes is:
mpdallexit
openmpi does not require starting a daemon or an mpd.hosts file.
A search using 9 processors can be run using the following command from your home directory:
mpiexec --machinefile mf -n 9 -wdir ~/Prospector/web/cgi-bin mssearchmpi.cgi -f ../../xml/bt_test.xml
The machinefile argument is used to specify a machine file (here called mf) which specifies how may processes to run on each node. This should add up to 9 in this case. Eg it could contain:
rodin04 slots=5 rodin05 slots=4
The wdir argument is used to specify the specify the working directory for mpiexec. In this case it is the directory containing the mssearchmpi.cgi file.
You first need to modify the .cshrc file in your home directory to add the web/cgi-bin directory to the PATH environment variable. A typical line you could add would be:
set path = ( ~/Prospector/web/cgi-bin $path )
Logout and login again to activate this change.
This cluster currently only has mpich2 installed and is running Redhat LINUX. There are 4 nodes called crick, franklin, watson and wilkins. When you ssh to plato.ucsf.edu you will be on one of these nodes. First connect to the crick node using the command:
ssh crick
The mpdboot command:
mpdboot --totalnum=2 --verbose --ifhn=crick-ci
The contents of the mpd.hosts file:
crick-ci watson-ci ifhn=watson-ci
The contents of the mf file:
crick-ci:9 watson-ci:8
Note that this is a slightly different format to the machine file described previously.
The mpiexec command:
mpiexec -machinefile mf -n 17 -wdir ~/Prospector/web/cgi-bin mssearchmpi.cgi -f ../../xml/bt_test.xml
The UCSF QB3 cluster is accessible via ssh at chef.compbio.ucsf.edu. The user login directories are shared by all nodes on the cluster.
If you need to compile the Prospector source code you need to ssh to the iq218 node using the command:
ssh iq218
As openmpi is available on the QB3 cluster there is no need to bother with mpich2. To compile the code for say version enter the following commands:
module load openmpi-x86_64 cd make clean make allclmpi
Short interactive jobs can also be run on the iq218 node to test the setup. Eg:
ssh iq218 module load openmpi-x86_64 cd Prospector/web/cgi-bin mpiexec -n 5 ./mssearchmpi.cgi -f ../../xml/bt_test.xml
To run a job on multiple nodes you need to submit a job to the SGE (Sun Grid Engine). To do this you first need to create a script such as the one shown below:
#!/bin/bash # #$ -S /bin/bash #$ -l arch=linux-x64 # Specify architecture, required #$ -l mem_free=1G # Memory usage, required. Note that this is per slot #$ -pe ompi 101 # Specify parallel environment and number of slots, required #$ -R yes # SGE host reservation, highly recommended #$ -V # Pass current environment to exec node, required #$ -cwd # Current working directory #$ -l h_rt=00:45:00 #-- runtime limit (see above; this requests 45 mins) module load openmpi-x86_64 mpirun -np $NSLOTS -wdir ~/Prospector/web/cgi-bin ./mssearchmpi.cgi -f ../../xml/bt_test.xml
As the script specifies the run directory of the mssearchmpi.cgi file then you can create a separate directory to hold these scripts and their output.
The line:
#$ -l mem_free=1G # Memory usage, required. Note that this is per slot
specifies the amount of memory required for each slot (mpi process), in this case 1 GByte.
The line:
#$ -pe ompi 101 # Specify parallel environment and number of slots, required
specifies the number of slots (mpi processes) to use, in this case 101.
The line:
#$ -l h_rt=00:45:00 #-- runtime limit (see above; this requests 45 mins)
specifies a runtime limit for the job, in this case 45 minutes. As your job will be aborted if the limit is exceeded you obviously need to leave some slack.
To submit the script enter the command:
qsub script
where script is the name of the script file.
You can submit a job on the chef node.
You can query the progress of the job using the command:
qstat
If the job has still not started the state column contains the value qw whereas if it is running it contains the value r. A finished job will not be in the table.
One of the columns in the qstat output contains a job-id. You can cancel a job with the command:
qdel -f job-id
A script has four output files:
script.e<job-id> script.o<job-id> script.pe<job-id> script.po<job-id>
The file script.o<job-id> can be inspected to check the progress of the search.
Search Compare can currently only run in single processor mode so it is run from the web/cgi-bin directory using a command line such as:
./searchCompare.cgi -f ../../xml/sc_test.xml
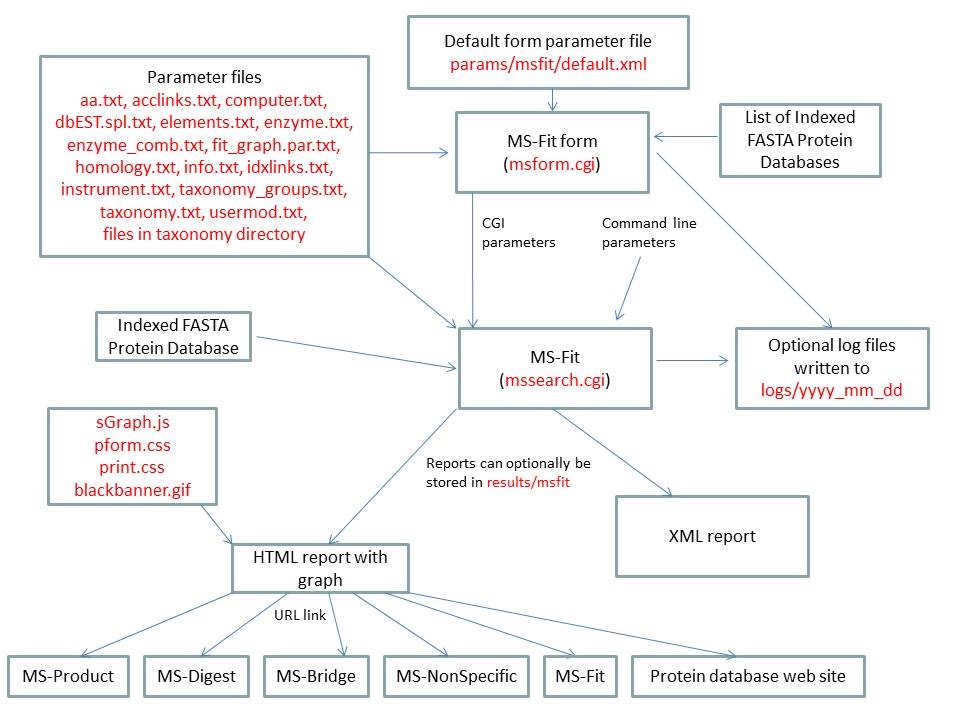
MS-Fit is run using the executable file cgi-bin/mssearch.cgi. The program can either be run from the command line or via a HTML form. The HTML form for running it is generated by the program cgi-bin/msform.cgi. The default form settings are stored in the file params/msfit/default.xml. The relevant parameter files are info.txt, aa.txt, acclinks.txt, computer.txt, dbEST.spl.txt (dbEST database searches only), elements.txt, enzyme.txt and enzyme_comb.txt, fit_graph.par.txt, homology.txt, idxlinks.txt, instrument.txt, taxonomy.txt and taxonomy_groups.txt (taxonomy searches only) and usermod.txt. The program also requires an indexed protein database produced by the FA-Index program. Possible output formats are HTML or XML. HTML reports make use of the Javascript file html/js/sGraph.js, the cascade stylesheet files html/pform.css and html/print.css and the image file html/images/bannerblack.gif. The HTML report contains URL links to the MS-Product, MS-Digest, MS-Bridge, MS-NonSpecific and MS-Fit programs and to external web sites which give more information about the proteins found in the search. If the output is written to a file rather than to the standard output then the file is stored in the directory results/mstag. The mssearch.cgi and msform.cgi programs can optionally write log files that are written to the directory logs/yyyy_mm_dd.
| MS-Fit | ||
|---|---|---|
| name | Default Value | Valid Values |
| search_name | "" | needs to be set to msfit |
| report_title | "" | text |
| output_type | HTML | HTML or XML |
| script | "" | text |
| script_type | "" | text |
| database | "" | valid prefixes: Genpept, gen, SwissProt, swp, Owl, owl, UniProt, Ludwignr, NCBInr, nr, dbEST, dbest, pdbEST, pdbest, IPI, ipi, DA, DN, PA, PN, pDA, pDN, Pdefault, Ddefault, pDdefault. User Protein is another possible selection. Multiple databases may be specified. |
| user_protein_sequence | "" | protein as a text string |
| instrument_name | "" | valid text strings from params/instrument.txt |
| dna_frame_translation | 3 | 6, 3, -3, 1, -1 |
| results_from_file | 0 | 0, 1 |
| results_input_dir | results_" + value of input_program_name parameter eg: results_msfit | directory name |
| input_program_name | msfit | msfit, mstag, mspattern, mshomology, msseq |
| input_filename | "" | text |
| indicies | "" | list of database indicies (integers) |
| accession_numbers | "" | list of database accession numbers |
| species | All | valid text strings from params/taxonomy.txt, params/taxonomy_groups.txt or All |
| names | None Defined | list of text strings |
| accession_nums | None Defined | list of text strings |
| species_names | None Defined | list of text strings |
| species_remove | 0 | 0, 1 |
| add_accession_numbers | None Defined | list of text strings |
| ms_prot_low_mass | 1000 | integer |
| ms_prot_high_mass | 100000 | integer |
| ms_full_mw_range | 0 | 0, 1 |
| low_pi | 3.0 | double |
| high_pi | 10.0 | double |
| full_pi_range | 0 | 0, 1 |
| sort_type | Score Sort | Score Sort MW Sort pI Sort |
| ms_report_homologous_proteins | Interesting | All, Interesting, None |
| enzyme | Trypsin | valid text strings from params/enzyme.txt or params/enzyme_comb.txt |
| missed_cleavages | 1 | integer |
| const_mod | None defined | valid text strings formed from the information in params/usermod.txt. |
| comment | "" | text |
| results_to_file | 0 | 0, 1 |
| output_dir | "results_" + value of search_name parameter ie: results_msfit | text |
| output_filename | "" | text |
| max_reported_hits | 50 | integer |
| detailed_report | 0 | 0,1 |
| display_graph | 0 | 0,1 |
| parent_mass_convert | monoisotopic | monoisotopic average Par(mi)Frag(av) Par(av)Frag(mi) |
| ms_parent_mass_tolerance | 0.5 | double |
| ms_parent_mass_tolerance_units | Da | Da, %, ppm, mmu |
| ms_parent_mass_systematic_error | 0.0 | double |
| parent_contaminant_masses | NULL | list of singly charged masses |
| average_to_mono_convert | NULL | list of 0's and 1's 0 = monoisotopic 1 = average |
| mod_AA | None Defined | Peptide N-terminal Gln to pyroGlu Oxidation of M Protein N-terminus Acetylated User Defined 1 Acrylamide Modified Cys |
| user1_name | "" | valid text strings defined in usermod.txt |
| min_parent_ion_matches | 1 | integer |
| min_matches | 5 | integer |
| mowse_on | 0 | 0, 1 |
| mowse_pfactor | 0.4 | double |
| data_source | Data Paste Area | List of Files, Upload Data From File, Data Paste Area |
| upload_data | "" | file name |
| data_directory | "" | directory name |
| data_files | NULL | list of file names |
| data_format | "" | M/Z Charge, M/Z Intensity Charge |
| data | NULL | See the data_format parameter. |
| ms_search_type | None Defined | valid text strings defined in params/homology.txt |
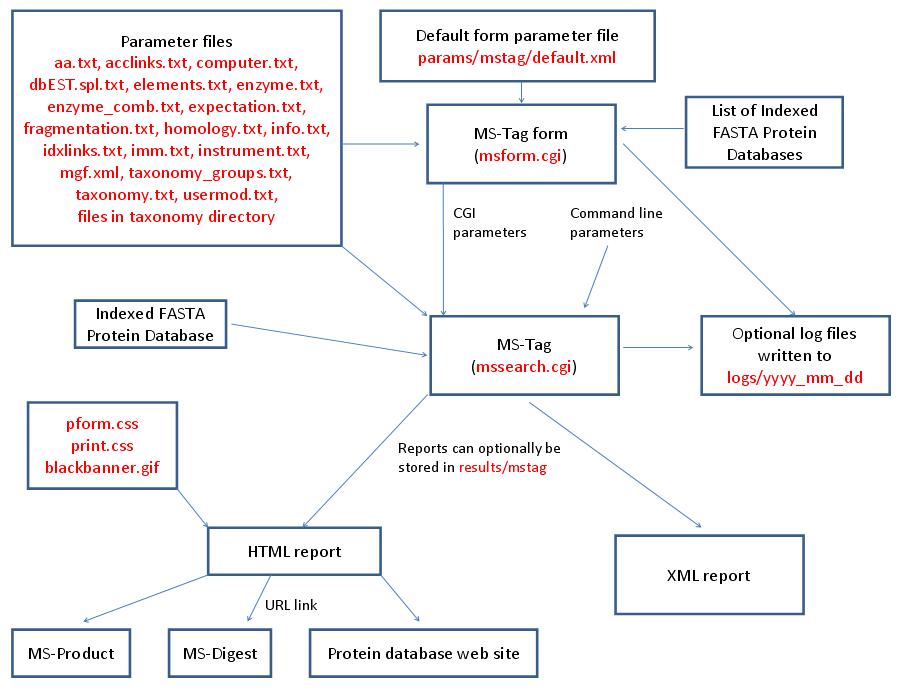
MS-Tag is run using the executable file cgi-bin/mssearch.cgi. The program can either be run from the command line or via a HTML form. The HTML form for running it is generated by the program cgi-bin/msform.cgi. The default form settings are stored in the file params/mstag/default.xml. The relevant parameter files are info.txt, aa.txt, acclinks.txt, computer.txt, dbEST.spl.txt (dbEST database searches only), elements.txt, enzyme.txt and enzyme_comb.txt (not required for No enzyme searches), expectation.txt, fragmentation.txt, homology.txt, idxlinks.txt, imm.txt, instrument.txt, mgf.xml, taxonomy.txt and taxonomy_groups.txt (taxonomy searches only) and usermod.txt. The program also requires an indexed protein database produced by the FA-Index program. Possible output formats are HTML or XML. HTML reports use the cascade stylesheet files html/pform.css and html/print.css and the image file html/images/bannerblack.gif. The HTML report contains URL links to the MS-Digest and MS-Product programs and to external web sites which give more information about the proteins found in the search. If the output is written to a file rather than to the standard output then the file is stored in the directory results/mstag. The mssearch.cgi and msform.cgi programs can optionally write log files that are written to the directory logs/yyyy_mm_dd.
| MS-Tag | ||
|---|---|---|
| name | Default Value | Valid Values |
| search_name | "" | needs to be set to mstag |
| report_title | "" | text |
| output_type | HTML | HTML or XML |
| script | "" | text |
| script_type | "" | text |
| database | "" | valid prefixes: Genpept, gen, SwissProt, swp, Owl, owl, UniProt, Ludwignr, NCBInr, nr, dbEST, dbest, pdbEST, pdbest, IPI, ipi, DA, DN, PA, PN, pDA, pDN, Pdefault, Ddefault, pDdefault. User Protein is another possible selection. Multiple databases may be specified. |
| user_protein_sequence | "" | protein as a text string |
| instrument_name | "" | valid text strings from params/instrument.txt |
| dna_frame_translation | 3 | 6, 3, -3, 1, -1 |
| results_from_file | 0 | 0, 1 |
| results_input_dir | results_" + value of input_program_name parameter eg: results_msfit | directory name |
| input_program_name | msfit | msfit, mstag, mspattern |
| input_filename | "" | text |
| indicies | "" | list of database indicies (integers) |
| accession_numbers | "" | list of database accession numbers |
| species | All | valid text strings from params/taxonomy.txt, params/taxonomy_groups.txt or All |
| names | None Defined | list of text strings |
| accession_nums | None Defined | list of text strings |
| species_names | None Defined | list of text strings |
| species_remove | 0 | 0, 1 |
| add_accession_numbers | None Defined | list of text strings |
| msms_prot_low_mass | 1000 | integer |
| msms_prot_high_mass | 100000 | integer |
| msms_full_mw_range | 0 | 0, 1 |
| low_pi | 3.0 | double |
| high_pi | 10.0 | double |
| full_pi_range | 0 | 0, 1 |
| enzyme | Trypsin | valid text strings from params/enzyme.txt or params/enzyme_comb.txt |
| allow_non_specific | at 0 termini | at 0 termini, at 1 termini, at 2 termini, at N termini, at C termini, N termini-1=D |
| missed_cleavages | 1 | integer |
| const_mod | None defined | valid text strings formed from the information in params/usermod.txt. |
| msms_mod_AA | None defined | valid text strings formed from the information in params/usermod.txt. |
| msms_max_modifications | 1 | integer |
| msms_max_peptide_permutations | "" | Left blank or non zero positive integer |
| comment | "" | text |
| expect_calc_method | None | None, Linear Tail Fit |
| bond_score | 0 | 0,1 |
| results_to_file | 0 | 0, 1 |
| output_dir | "results_" + value of search_name parameter ie: results_mstag | text |
| output_filename | "" | text |
| max_reported_hits | 50 | integer |
| max_hits | 200 | integer |
| parent_mass_convert | monoisotopic | monoisotopic average Par(mi)Frag(av) Par(av)Frag(mi) |
| ms_parent_mass_tolerance | 0.5 | double |
| ms_parent_mass_tolerance_units | Da | Da, %, ppm, mmu |
| ms_parent_mass_systematic_error | 0.0 | double |
| data_source | Data Paste Area | List of Files, Upload Data From File, Data Paste Area |
| upload_data | "" | file name |
| data_directory | "" | directory name |
| data_files | NULL | list of file names |
| data_format | "" | M/Z Charge, M/Z Intensity Charge |
| data | NULL | See the data_format parameter. For MS-Tag the first line contains the m/z and optionally the charge of the parent ion. |
| mod_start_nominal | 0 | integer |
| mod_end_nominal | 0 | integer |
| mod_defect | 0.0 | double |
| mod_comp_ion | None defined | A,C,D,E,F,G,H,I,K,L,M,N,P,Q,R,S,T,V,W,Y |
| mod_n_term | 0 | 0, 1 |
| mod_c_term | 0 | 0, 1 |
| mod_neutral_loss | 0 | 0, 1 |
| mod_uncleaved | 0 | 0, 1 |
| msms_pk_filter | Max MSMS Pks | Max MSMS Pks, Max MSMS Pks / 100 Da or Unprocessed MSMS |
| msms_search_type | None Defined | valid text strings defined in params/homology.txt |
| it | None Defined | a,a-H2O,a-NH3,a-H3PO4, b,b-H2O,b-NH3,b+H2O, b-H3PO4,b-SOCH4, y,y-H2O,y-NH3,y-H3PO4,y-SOCH4, MH+,B,c-1,c,c+1,c+2,x,Y,z,z+1,z+2,z+3,n,h,P,S,I,N,C |
| fragment_masses_tolerance | 1.0 | double |
| fragment_masses_tolerance_units | Da | Da, %, ppm, mmu |
| comp_ion | None Defined | regular expression |
| composition_search | 0 | 0, 1 |
| exclude_flag | 0 | 0, 1 |
| aa_exclude | "" | text string containing the following characters A,C,D,E,F,G,H,I,K,L,M,N,P,Q,R,S,T,V,W,Y |
| aa_add | "" | d,e,f,g,h,i,j,k,l,m,s,t,y,z |
| user_aa_composition | C2 H3 N1 O1 | Elemental formula of the form Cx Hy Oz etc where x, y and z are integers. Elements defined in params/elements.txt can be used. |
| user_aa_2_composition | "" | Elemental formula of the form Cx Hy Oz etc where x, y and z are integers. Elements defined in params/elements.txt can be used. |
| user_aa_3_composition | "" | Elemental formula of the form Cx Hy Oz etc where x, y and z are integers. Elements defined in params/elements.txt can be used. |
| user_aa_4_composition | "" | Elemental formula of the form Cx Hy Oz etc where x, y and z are integers. Elements defined in params/elements.txt can be used. |
| user_aa_5_composition | "" | Elemental formula of the form Cx Hy Oz etc where x, y and z are integers. Elements defined in params/elements.txt can be used. |
| user_aa_6_composition | "" | Elemental formula of the form Cx Hy Oz etc where x, y and z are integers. Elements defined in params/elements.txt can be used. |
| user_aa_7_composition | "" | Elemental formula of the form Cx Hy Oz etc where x, y and z are integers. Elements defined in params/elements.txt can be used. |
| user_aa_8_composition | "" | Elemental formula of the form Cx Hy Oz etc where x, y and z are integers. Elements defined in params/elements.txt can be used. |
| user_aa_9_composition | "" | Elemental formula of the form Cx Hy Oz etc where x, y and z are integers. Elements defined in params/elements.txt can be used. |
| regular_expression | . | regular expression |
| use_instrument_ion_types | 0 | 0,1 |
| score_histogram_only | 0 | 0,1. The option is not currently available on the form but could be set in the mstag/default.xml file. If set then MS-Tag will just produce a score histogram rather than the standard report. |
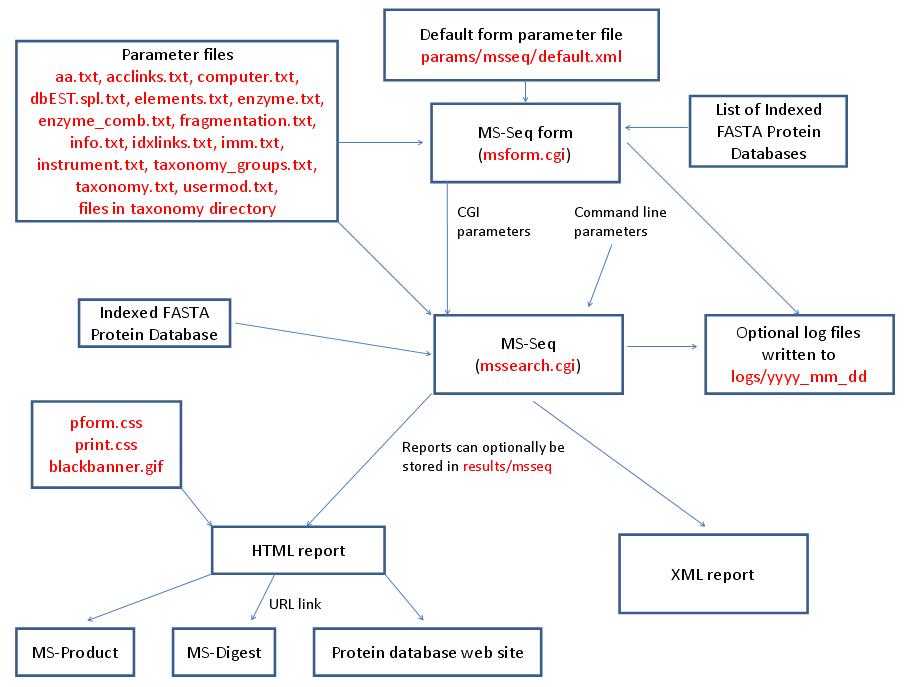
MS-Seq is run using the executable file cgi-bin/mssearch.cgi. The program can either be run from the command line or via a HTML form. The HTML form for running it is generated by the program cgi-bin/msform.cgi. The default form settings are stored in the file params/msseq/default.xml. The relevant parameter files are info.txt, aa.txt, acclinks.txt, computer.txt, dbEST.spl.txt (dbEST database searches only), elements.txt, enzyme.txt and enzyme_comb.txt (not required for No enzyme searches), fragmentation.txt, idxlinks.txt, imm.txt, instrument.txt, taxonomy.txt and taxonomy_groups.txt (taxonomy searches only) and usermod.txt. The program also requires an indexed protein database produced by the FA-Index program. Possible output formats are HTML or XML. HTML reports use the cascade stylesheet files html/pform.css and html/print.css and the image file html/images/bannerblack.gif. The HTML report contains URL links to the MS-Digest and MS-Product programs and to external web sites which give more information about the proteins found in the search. If the output is written to a file rather than to the standard output then the file is stored in the directory results/msseq. The mssearch.cgi and msform.cgi programs can optionally write log files that are written to the directory logs/yyyy_mm_dd.
| MS-Seq | ||
|---|---|---|
| name | Default Value | Valid Values |
| search_name | "" | text which can be part of a filename. |
| report_title | "" | needs to be set to msseq |
| output_type | HTML | HTML or XML |
| script | "" | text |
| script_type | "" | text |
| database | "" | valid prefixes: Genpept, gen, SwissProt, swp, Owl, owl, UniProt, Ludwignr, NCBInr, nr, dbEST, dbest, pdbEST, pdbest, IPI, ipi, DA, DN, PA, PN, pDA, pDN, Pdefault, Ddefault, pDdefault. User Protein is another possible selection. Multiple databases may be specified. |
| user_protein_sequence | "" | protein as a text string |
| instrument_name | "" | valid text strings from params/instrument.txt |
| dna_frame_translation | 3 | 6, 3, -3, 1, -1 |
| results_from_file | 0 | 0, 1 |
| results_input_dir | results_" + value of input_program_name parameter eg: results_msfit | directory name |
| input_program_name | msfit | msfit, mstag, mspattern, mshomology, msseq |
| input_filename | "" | text |
| indicies | "" | list of database indicies (integers) |
| accession_numbers | "" | list of database accession numbers |
| species | All | valid text strings from params/taxonomy.txt, params/taxonomy_groups.txt or All |
| names | None Defined | list of text strings |
| accession_nums | None Defined | list of text strings |
| species_names | None Defined | list of text strings |
| species_remove | 0 | 0, 1 |
| add_accession_numbers | None Defined | list of text strings |
| msms_prot_low_mass | 1000 | integer |
| msms_prot_high_mass | 100000 | integer |
| msms_full_mw_range | 0 | 0, 1 |
| low_pi | 3.0 | double |
| high_pi | 10.0 | double |
| full_pi_range | 0 | 0, 1 |
| enzyme | Trypsin | valid text strings from params/enzyme.txt or params/enzyme_comb.txt |
| allow_non_specific | at 0 termini | at 0 termini, at 1 termini, at 2 termini, at N termini, at C termini, N termini-1=D |
| missed_cleavages | 1 | integer |
| const_mod | None defined | valid text strings formed from the information in params/usermod.txt. |
| comment | "" | text |
| results_to_file | 0 | 0, 1 |
| output_dir | "results_" + value of search_name parameter ie: results_msseq | text |
| output_filename | "" | text |
| max_reported_hits | 50 | integer |
| max_hits | 200 | integer |
| parent_mass_convert | monoisotopic | monoisotopic average Par(mi)Frag(av) Par(av)Frag(mi) |
| ms_parent_mass_tolerance | 0.5 | double |
| ms_parent_mass_tolerance_units | Da | Da, %, ppm, mmu |
| ms_parent_mass_systematic_error | 0.0 | double |
| fragment_masses_tolerance | 1.0 | double |
| fragment_masses_tolerance_units | Da | Da, %, ppm, mmu |
| data_format | "" | M/Z Charge, M/Z Intensity Charge |
| data | NULL | See the data_format parameter. For MS-Tag the first line contains the m/z and optionally the charge of the parent ion. |
| msms_search_type | None Defined | no errors, high mass error, low mass error, middle masses error, parent mass |
| ion_type | b | a,b,c,y |
| fragment_mass_tolerance | 1.0 | double |
| comp_ion | None Defined | regular expression |
| composition_search | 0 | 0, 1 |
| composition_exclude | "" | ACDEFGHIKLMNPQRSTVWY |
| regular_expression | . | regular expression |
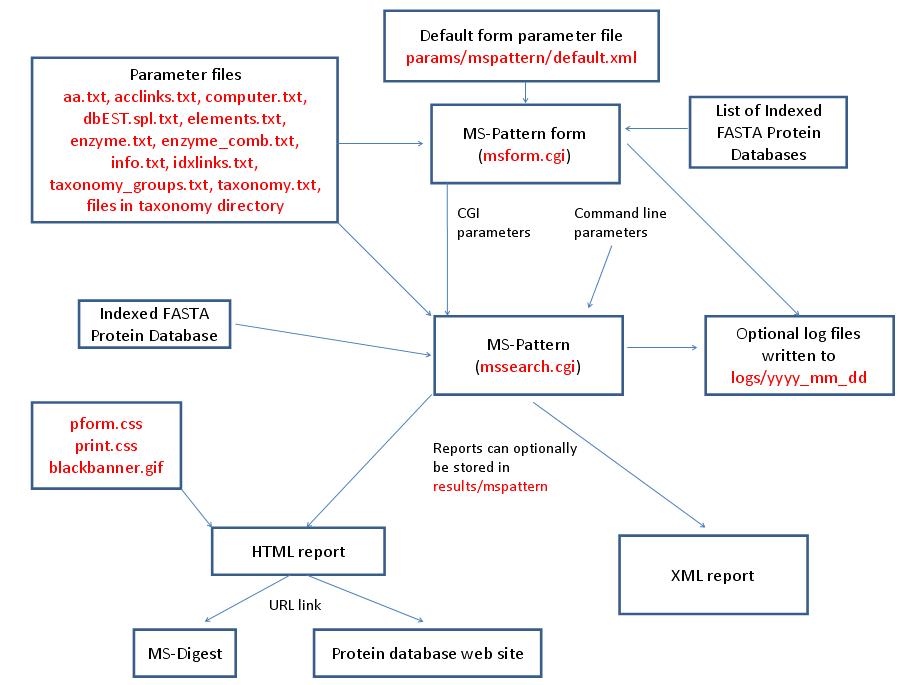
MS-Pattern is run using the executable file cgi-bin/mssearch.cgi. The program can either be run from the command line or via a HTML form. The HTML form for running it is generated by the program cgi-bin/msform.cgi. The default form settings are stored in the file params/mspattern/default.xml. The relevant parameter files are info.txt, aa.txt, acclinks.txt, computer.txt, dbEST.spl.txt (dbEST database searches only), elements.txt, enzyme.txt and enzyme_comb.txt (not required for No enzyme searches), idxlinks.txt and taxonomy.txt and taxonomy_groups.txt (taxonomy searches only). The program also requires an indexed protein database produced by the FA-Index program. Possible output formats are HTML or XML. HTML reports use the cascade stylesheet files html/pform.css and html/print.css and the image file html/images/bannerblack.gif. The HTML report contains URL links to the MS-Digest program and to external web sites which give more information about the proteins found in the search. If the output is written to a file rather than to the standard output then the file is stored in the directory results/mspattern. The mssearch.cgi and msform.cgi programs can optionally write log files that are written to the directory logs/yyyy_mm_dd.
| MS-Pattern | ||
|---|---|---|
| name | Default Value | Valid Values |
| search_name | "" | text which can be part of a filename. |
| report_title | "" | needs to be set to mspattern |
| output_type | HTML | HTML or XML |
| script | "" | text |
| script_type | "" | text |
| database | "" | valid prefixes: Genpept, gen, SwissProt, swp, Owl, owl, UniProt, Ludwignr, NCBInr, nr, dbEST, dbest, pdbEST, pdbest, IPI, ipi, DA, DN, PA, PN, pDA, pDN, Pdefault, Ddefault, pDdefault. User Protein is another possible selection. Multiple databases may be specified. |
| user_protein_sequence | "" | protein as a text string |
| dna_frame_translation | 3 | 6, 3, -3, 1, -1 |
| results_from_file | 0 | 0, 1 |
| results_input_dir | results_" + value of input_program_name parameter eg: results_msfit | directory name |
| input_program_name | msfit | msfit, mstag, mspattern, mshomology, msseq |
| input_filename | "" | text |
| indicies | "" | list of database indicies (integers) |
| accession_numbers | "" | list of database accession numbers |
| species | All | valid text strings from params/taxonomy.txt, params/taxonomy_groups.txt or All |
| names | None Defined | list of text strings |
| accession_nums | None Defined | list of text strings |
| species_names | None Defined | list of text strings |
| species_remove | 0 | 0, 1 |
| add_accession_numbers | None Defined | list of text strings |
| prot_low_mass | 1000 | integer |
| prot_high_mass | 100000 | integer |
| full_mw_range | 0 | 0, 1 |
| low_pi | 3.0 | double |
| high_pi | 10.0 | double |
| full_pi_range | 0 | 0, 1 |
| enzyme | Trypsin | valid text strings from params/enzyme.txt or params/enzyme_comb.txt |
| comment | "" | text |
| results_to_file | 0 | 0, 1 |
| output_dir | "results_" + value of search_name parameter ie: results_mspattern | text |
| output_filename | "" | text |
| max_reported_hits | 50 | integer |
| pre_search_only | 0 | 0,1 |
| regular_expression | . | regular expression |
| possible_sequences | NULL | list of peptides |
| max_aa_substitutions | 0 | integer |
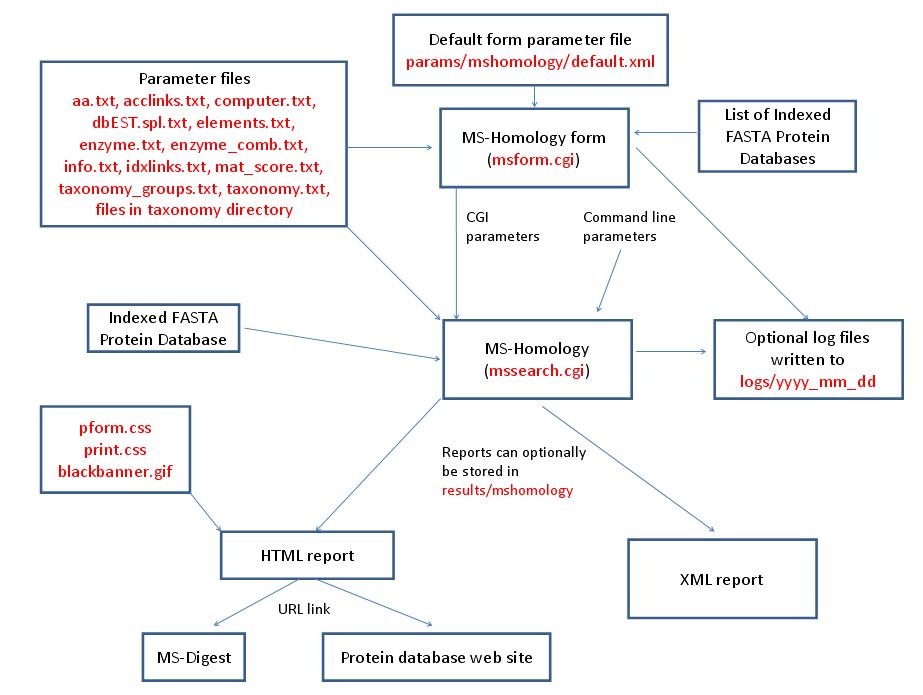
MS-Homology is run using the executable file cgi-bin/mssearch.cgi. The program can either be run from the command line or via a HTML form. The HTML form for running it is generated by the program cgi-bin/msform.cgi. The default form settings are stored in the file params/mshomology/default.xml. The relevant parameter files are info.txt, aa.txt, acclinks.txt, computer.txt, dbEST.spl.txt (dbEST database searches only), elements.txt, enzyme.txt and enzyme_comb.txt (not required for No enzyme searches), idxlinks.txt, mat_score.txt and taxonomy.txt and taxonomy_groups.txt (taxonomy searches only). The program also requires an indexed protein database produced by the FA-Index program. Possible output formats are HTML or XML. HTML reports use the cascade stylesheet files html/pform.css and html/print.css and the image file html/images/bannerblack.gif. The HTML report contains URL links to the MS-Digest program and to external web sites which give more information about the proteins found in the search. If the output is written to a file rather than to the standard output then the file is stored in the directory results/mshomology. The mssearch.cgi and msform.cgi programs can optionally write log files that are written to the directory logs/yyyy_mm_dd.
| MS-Homology | ||
|---|---|---|
| name | Default Value | Valid Values |
| search_name | "" | text which can be part of a filename. |
| report_title | "" | needs to be set to mshomology |
| output_type | HTML | HTML, XML or Tab delimited text |
| script | "" | text |
| script_type | "" | text |
| database | "" | valid prefixes: Genpept, gen, SwissProt, swp, Owl, owl, UniProt, Ludwignr, NCBInr, nr, dbEST, dbest, pdbEST, pdbest, IPI, ipi, DA, DN, PA, PN, pDA, pDN, Pdefault, Ddefault, pDdefault. User Protein is another possible selection. Multiple databases may be specified. |
| user_protein_sequence | "" | protein as a text string |
| dna_frame_translation | 3 | 6, 3, -3, 1, -1 |
| results_from_file | 0 | 0, 1 |
| results_input_dir | results_" + value of input_program_name parameter eg: results_msfit | directory name |
| input_program_name | msfit | msfit, mstag, mspattern, mshomology, msseq |
| input_filename | "" | text |
| indicies | "" | list of database indicies (integers) |
| accession_numbers | "" | list of database accession numbers |
| species | All | valid text strings from params/taxonomy.txt, params/taxonomy_groups.txt or All |
| names | None Defined | list of text strings |
| accession_nums | None Defined | list of text strings |
| species_names | None Defined | list of text strings |
| species_remove | 0 | 0, 1 |
| add_accession_numbers | None Defined | list of text strings |
| prot_low_mass | 1000 | integer |
| prot_high_mass | 100000 | integer |
| full_mw_range | 0 | 0, 1 |
| low_pi | 3.0 | double |
| high_pi | 10.0 | double |
| full_pi_range | 0 | 0, 1 |
| enzyme | Trypsin | valid text strings from params/enzyme.txt or params/enzyme_comb.txt |
| comment | "" | text |
| previous_aa | 1 | non-zero positive integer |
| next_aa | 1 | non-zero positive integer |
| results_to_file | 0 | 0, 1 |
| output_dir | "results_" + value of search_name parameter ie: results_mshomology | text |
| output_filename | "" | text |
| max_reported_hits | 50 | integer |
| parent_mass_convert | monoisotopic | monoisotopic average Par(mi)Frag(av) Par(av)Frag(mi) |
| fragment_masses_tolerance | 1.0 | double |
| fragment_masses_tolerance_units | Da | Da, %, ppm, mmu |
| min_matches | 5 | integer |
| possible_sequences | NULL | list of peptides |
| score_matrix | 0 | integer |
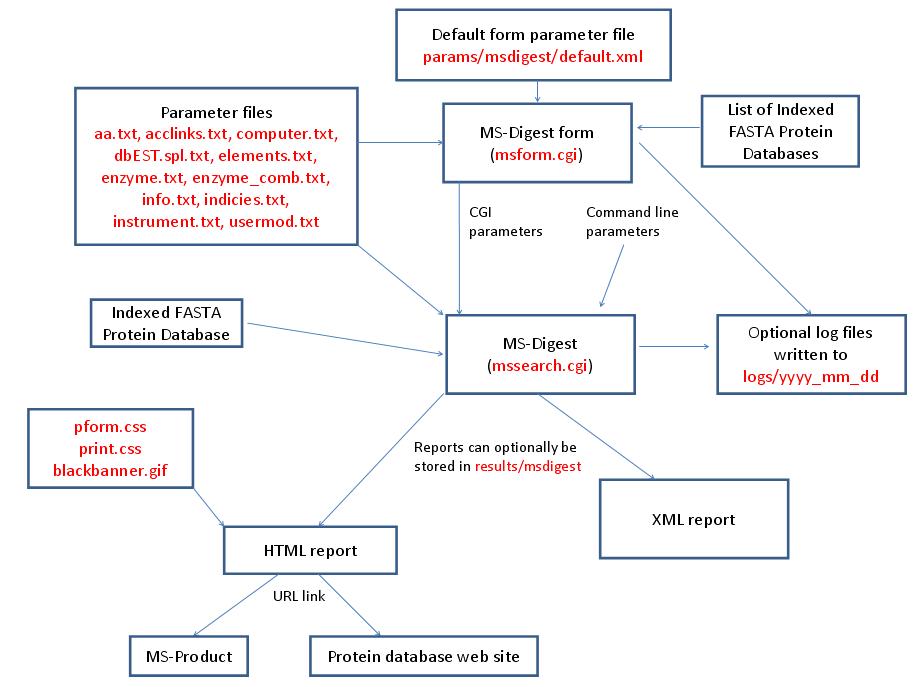
MS-Digest is run using the executable file cgi-bin/mssearch.cgi. The program can either be run from the command line or via a HTML form. The HTML form for running it is generated by the program cgi-bin/msform.cgi. The default form settings are stored in the file params/msdigest/default.xml. The relevant parameter files are info.txt, aa.txt, acclinks.txt, computer.txt, dbEST.spl.txt (dbEST database searches only), elements.txt, enzyme.txt and enzyme_comb.txt, indicies.txt, instrument.txt and usermod.txt. The program can either use entries from an indexed protein database produced by the FA-Index program or protein sequences entered as one of the program parameters. Possible output formats are HTML or XML. HTML reports makes use of the the cascade stylesheet files html/pform.css and html/print.css and the image file html/images/bannerblack.gif. The HTML report contains URL links to the MS-Product program and to external web sites which give more information about the proteins found in the search. If the output is written to a file rather than to the standard output then the file is stored in the directory results/msdigest. The mssearch.cgi and msform.cgi programs can optionally write log files that are written to the directory logs/yyyy_mm_dd.
| MS-Digest | ||
|---|---|---|
| name | Default Value | Valid Values |
| search_name | "" | text which can be part of a filename. |
| report_title | "" | needs to be set to msdigest |
| output_type | HTML | HTML, XML or Tab delimited text |
| script | "" | text |
| script_type | "" | text |
| database | "" | valid prefixes: Genpept, gen, SwissProt, swp, Owl, owl, UniProt, Ludwignr, NCBInr, nr, dbEST, dbest, pdbEST, pdbest, IPI, ipi, DA, DN, PA, PN, pDA, pDN, Pdefault, Ddefault, pDdefault. User Protein is another possible selection. |
| instrument_name | "" | valid text strings from params/instrument.txt |
| enzyme | Trypsin | valid text strings from params/enzyme.txt or params/enzyme_comb.txt |
| missed_cleavages | 1 | integer |
| const_mod | None defined | valid text strings formed from the information in params/usermod.txt. |
| end_terminus | 0 | 0, 1 |
| stripping_terminus | N | N, C |
| start_strip | 2 | integer |
| end_strip | 4 | integer |
| results_to_file | 0 | 0, 1 |
| output_dir | "results_" + value of search_name parameter ie: results_msdigest | text |
| output_filename | "" | text |
| parent_mass_convert | monoisotopic | monoisotopic average Par(mi)Frag(av) Par(av)Frag(mi) |
| mod_AA | None defined | valid text strings formed from the information in params/usermod.txt. |
| bull_breese | 0 | 0,1 |
| hplc_index | 0 | 0,1 |
| comp_ion | None Defined | regular expression |
| comp_mask_type | AND | AND or OR |
| user_aa_composition | C2 H3 N1 O1 | Elemental formula of the form Cx Hy Oz etc where x, y and z are integers. Elements defined in params/elements.txt can be used. |
| user_aa_2_composition | "" | Elemental formula of the form Cx Hy Oz etc where x, y and z are integers. Elements defined in params/elements.txt can be used. |
| user_aa_3_composition | "" | Elemental formula of the form Cx Hy Oz etc where x, y and z are integers. Elements defined in params/elements.txt can be used. |
| user_aa_4_composition | "" | Elemental formula of the form Cx Hy Oz etc where x, y and z are integers. Elements defined in params/elements.txt can be used. |
| user_aa_5_composition | "" | Elemental formula of the form Cx Hy Oz etc where x, y and z are integers. Elements defined in params/elements.txt can be used. |
| user_aa_6_composition | "" | Elemental formula of the form Cx Hy Oz etc where x, y and z are integers. Elements defined in params/elements.txt can be used. |
| user_aa_7_composition | "" | Elemental formula of the form Cx Hy Oz etc where x, y and z are integers. Elements defined in params/elements.txt can be used. |
| user_aa_8_composition | "" | Elemental formula of the form Cx Hy Oz etc where x, y and z are integers. Elements defined in params/elements.txt can be used. |
| user_aa_9_composition | "" | Elemental formula of the form Cx Hy Oz etc where x, y and z are integers. Elements defined in params/elements.txt can be used. |
| dna_reading_frame | 1 | 1, 2, 3, 4, 5, 6 |
| open_reading_frame | 1 | non-zero positive integer |
| user_protein_sequence | "" | protein as a text string |
| report_mult_charge | 0 | 0, 1 |
| hide_html_links | 0 | 0, 1 |
| separate_proteins | 0 | 0, 1 |
| hide_protein_sequence | 0 | 0, 1 |
| access_method | Index Number | Index Number Accession Number |
| entry_data | "" | See manual |
| index_num | 80707 | integer |
| accession_num | L39370 | text |
| min_digest_fragment_mass | 500.0 | double |
| max_digest_fragment_mass | 4000.0 | double |
| min_digest_fragment_length | 5 | integer |
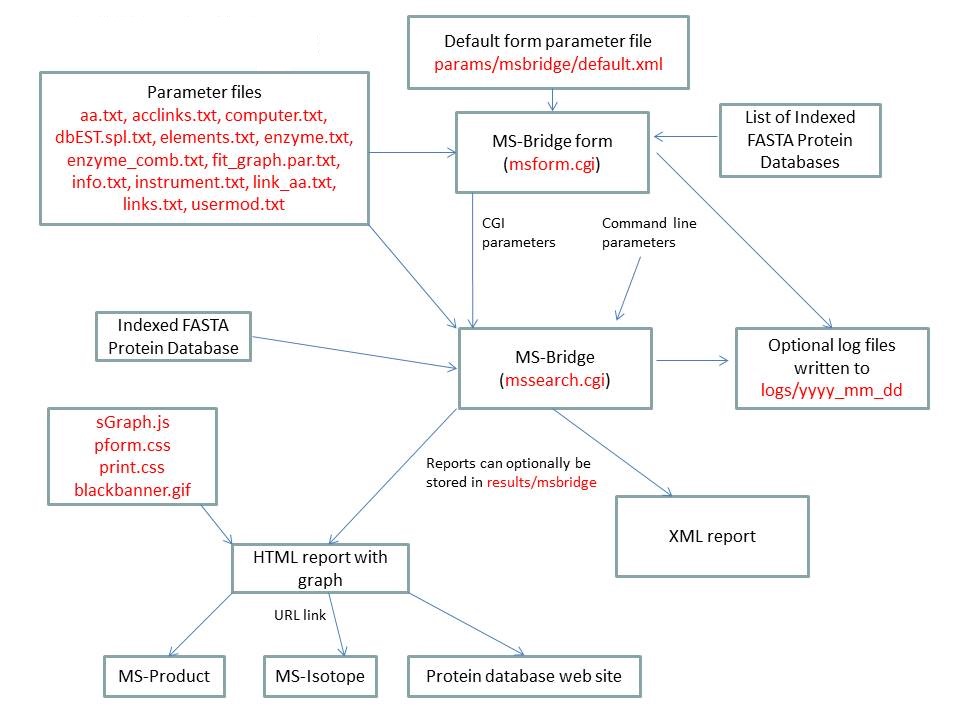
MS-Bridge is run using the executable file cgi-bin/mssearch.cgi. The program can either be run from the command line or via a HTML form. The HTML form for running it is generated by the program cgi-bin/msform.cgi. The default form settings are stored in the file params/msbridge/default.xml. The relevant parameter files are info.txt, aa.txt, acclinks.txt, computer.txt, dbEST.spl.txt (dbEST database searches only), elements.txt, enzyme.txt and enzyme_comb.txt, fit_graph.par.txt, instrument.txt, link_aa.txt, links.txt and usermod.txt. The program can either use entries from an indexed protein database produced by the FA-Index program or protein sequences entered as one of the program parameters. Possible output formats are HTML or XML. HTML reports make use of the Javascript file html/js/sGraph.js, the cascade stylesheet files html/pform.css and html/print.css and the image file html/images/bannerblack.gif. The HTML report contains URL links to the MS-Product and MS-Isotope programs and to external web sites which give more information about the proteins found in the search. If the output is written to a file rather than to the standard output then the file is stored in the directory results/mstag. The mssearch.cgi and msform.cgi programs can optionally write log files that are written to the directory logs/yyyy_mm_dd.
| MS-Bridge | ||
|---|---|---|
| name | Default Value | Valid Values |
| search_name | "" | text which can be part of a filename. |
| report_title | "" | needs to be set to msbridge |
| output_type | HTML | HTML or XML |
| script | "" | text |
| script_type | "" | text |
| database | "" | valid prefixes: Genpept, gen, SwissProt, swp, Owl, owl, UniProt, Ludwignr, NCBInr, nr, dbEST, dbest, pdbEST, pdbest, IPI, ipi, DA, DN, PA, PN, pDA, pDN, Pdefault, Ddefault, pDdefault. User Protein is another possible selection. |
| instrument_name | "" | valid text strings from params/instrument.txt |
| enzyme | Trypsin | valid text strings from params/enzyme.txt or params/enzyme_comb.txt |
| missed_cleavages | 1 | integer |
| const_mod | None defined | valid text strings formed from the information in params/usermod.txt. |
| end_terminus | 0 | 0, 1 |
| stripping_terminus | N | N, C |
| start_strip | 2 | integer |
| end_strip | 4 | integer |
| results_to_file | 0 | 0, 1 |
| output_dir | "results_" + value of search_name parameter ie: results_msbridge | text |
| output_filename | "" | text |
| parent_mass_convert | monoisotopic | monoisotopic average Par(mi)Frag(av) Par(av)Frag(mi) |
| ms_parent_mass_tolerance | 0.5 | double |
| ms_parent_mass_tolerance_units | Da | Da, %, ppm, mmu |
| ms_parent_mass_systematic_error | 0.0 | double |
| parent_contaminant_masses | NULL | list of singly charged masses |
| mod_AA | None defined | valid text strings formed from the information in params/usermod.txt. |
| link_search_type | Xlink:Dehydro (C) | valid text strings defined in links.txt |
| max_link_molecules | 5 | integer |
| link_aa | "" | string of the form C->C where the amino acids on each side of the cross-link are separated by ->. If there are multiple possibilities they should be separated by commas (eg. K,R->K,R). If one possibility is the protein N or C-terminus use this notation: K,Protein N-term->Q. |
| bridge_composition | "" | Elemental formula of the form Cx Hy Oz etc where x, y and z are integers. Elements defined in params/elements.txt can be used. |
| mod_1_label | "" | string |
| aa_modified_1 | "" | An amino acid code or string such as Protein N-term. |
| mod_1_composition | "" | Elemental formula of the form Cx Hy Oz etc where x, y and z are integers. Elements defined in params/elements.txt can be used. |
| mod_2_label | "" | string |
| aa_modified_2 | "" | An amino acid code or string such as Protein N-term. |
| mod_2_composition | "" | Elemental formula of the form Cx Hy Oz etc where x, y and z are integers. Elements defined in params/elements.txt can be used. |
| mod_3_label | "" | string |
| aa_modified_3 | "" | An amino acid code or string such as Protein N-term. |
| mod_3_composition | "" | Elemental formula of the form Cx Hy Oz etc where x, y and z are integers. Elements defined in params/elements.txt can be used. |
| mod_4_label | "" | string |
| aa_modified_4 | "" | An amino acid code or string such as Protein N-term. |
| mod_4_composition | "" | Elemental formula of the form Cx Hy Oz etc where x, y and z are integers. Elements defined in params/elements.txt can be used. |
| mod_5_label | "" | string |
| aa_modified_5 | "" | An amino acid code or string such as Protein N-term. |
| mod_5_composition | "" | Elemental formula of the form Cx Hy Oz etc where x, y and z are integers. Elements defined in params/elements.txt can be used. |
| mod_6_label | "" | string |
| aa_modified_6 | "" | An amino acid code or string such as Protein N-term. |
| mod_6_composition | "" | Elemental formula of the form Cx Hy Oz etc where x, y and z are integers. Elements defined in params/elements.txt can be used. |
| data_source | Data Paste Area | List of Files, Upload Data From File, Data Paste Area |
| upload_data | "" | file name |
| data_format | "" | M/Z Charge, M/Z Intensity Charge |
| data | NULL | See the data_format parameter. |
| comp_ion | None Defined | regular expression |
| comp_mask_type | AND | AND or OR |
| user_aa_composition | C2 H3 N1 O1 | Elemental formula of the form Cx Hy Oz etc where x, y and z are integers. Elements defined in params/elements.txt can be used. |
| user_aa_2_composition | "" | Elemental formula of the form Cx Hy Oz etc where x, y and z are integers. Elements defined in params/elements.txt can be used. |
| user_aa_3_composition | "" | Elemental formula of the form Cx Hy Oz etc where x, y and z are integers. Elements defined in params/elements.txt can be used. |
| user_aa_4_composition | "" | Elemental formula of the form Cx Hy Oz etc where x, y and z are integers. Elements defined in params/elements.txt can be used. |
| user_aa_5_composition | "" | Elemental formula of the form Cx Hy Oz etc where x, y and z are integers. Elements defined in params/elements.txt can be used. |
| user_aa_6_composition | "" | Elemental formula of the form Cx Hy Oz etc where x, y and z are integers. Elements defined in params/elements.txt can be used. |
| user_aa_7_composition | "" | Elemental formula of the form Cx Hy Oz etc where x, y and z are integers. Elements defined in params/elements.txt can be used. |
| user_aa_8_composition | "" | Elemental formula of the form Cx Hy Oz etc where x, y and z are integers. Elements defined in params/elements.txt can be used. |
| user_aa_9_composition | "" | Elemental formula of the form Cx Hy Oz etc where x, y and z are integers. Elements defined in params/elements.txt can be used. |
| dna_reading_frame | 1 | 1, 2, 3, 4, 5, 6 |
| open_reading_frame | 1 | non-zero positive integer |
| user_protein_sequence | "" | protein as a text string |
| separate_proteins | 0 | 0, 1 |
| hide_protein_sequence | 0 | 0, 1 |
| access_method | Index Number | Index Number Accession Number |
| entry_data | "" | See manual |
| index_num | 80707 | integer |
| accession_num | L39370 | text |
| min_digest_fragment_mass | 500.0 | double |
| max_digest_fragment_mass | 4000.0 | double |
| min_digest_fragment_length | 5 | integer |
MS-NonSpecific is run using the executable file cgi-bin/mssearch.cgi. The program can either be run from the command line or via a HTML form. The HTML form for running it is generated by the program cgi-bin/msform.cgi. The default form settings are stored in the file params/msnonspecific/default.xml. The relevant parameter files are info.txt, aa.txt, acclinks.txt, computer.txt, dbEST.spl.txt (dbEST database searches only), elements.txt, fit_graph.par.txt, instrument.txt and usermod.txt. The program can either use entries from an indexed protein database produced by the FA-Index program or protein sequences entered as one of the program parameters. Possible output formats are HTML or XML. HTML reports make use of the Javascript file html/js/sGraph.js, the cascade stylesheet files html/pform.css and html/print.css and the image file html/images/bannerblack.gif. The HTML report contains URL links to the MS-Product program and to external web sites which give more information about the proteins found in the search. If the output is written to a file rather than to the standard output then the file is stored in the directory results/msnonspecific. The mssearch.cgi and msform.cgi programs can optionally write log files that are written to the directory logs/yyyy_mm_dd.
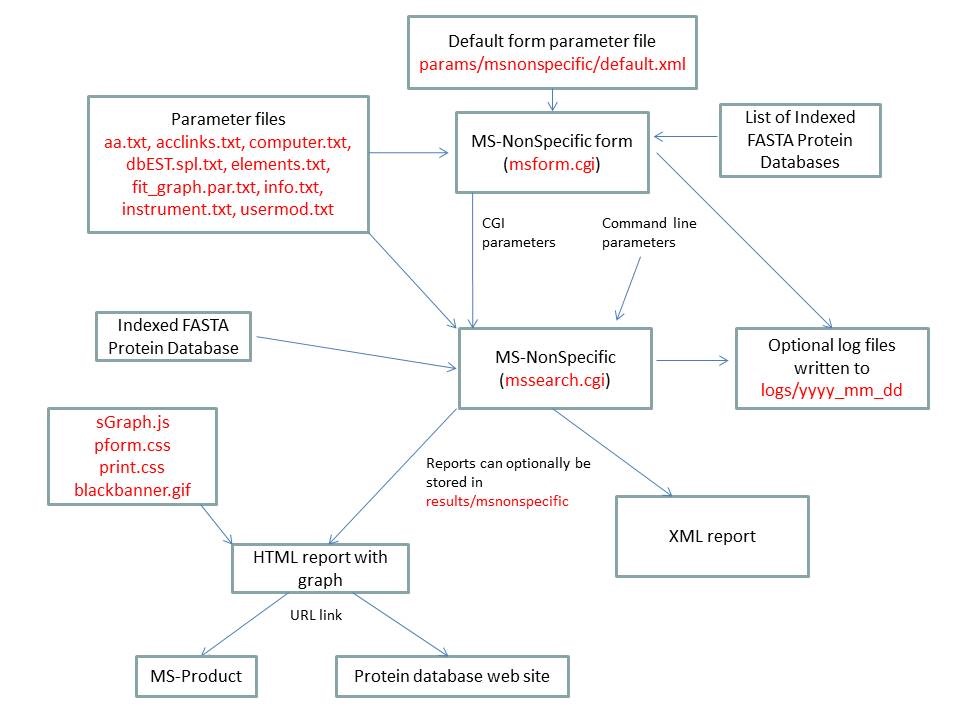
| MS-NonSpecific | ||
|---|---|---|
| name | Default Value | Valid Values |
| search_name | "" | text which can be part of a filename. |
| report_title | "" | needs to be set to msnonspecific |
| output_type | HTML | HTML or XML |
| script | "" | text |
| script_type | "" | text |
| database | "" | valid prefixes: Genpept, gen, SwissProt, swp, Owl, owl, UniProt, Ludwignr, NCBInr, nr, dbEST, dbest, pdbEST, pdbest, IPI, ipi, DA, DN, PA, PN, pDA, pDN, Pdefault, Ddefault, pDdefault. User Protein is another possible selection. |
| instrument_name | "" | valid text strings from params/instrument.txt |
| const_mod | None defined | valid text strings formed from the information in params/usermod.txt. |
| results_to_file | 0 | 0, 1 |
| output_dir | "results_" + value of search_name parameter ie: results_msnonspecific | text |
| output_filename | "" | text |
| parent_mass_convert | monoisotopic | monoisotopic average Par(mi)Frag(av) Par(av)Frag(mi) |
| ms_parent_mass_tolerance | 0.5 | double |
| ms_parent_mass_tolerance_units | Da | Da, %, ppm, mmu |
| ms_parent_mass_systematic_error | 0.0 | double |
| parent_contaminant_masses | NULL | list of singly charged masses |
| data_source | Data Paste Area | List of Files, Upload Data From File, Data Paste Area |
| upload_data | "" | file name |
| data_format | "" | M/Z Charge, M/Z Intensity Charge |
| data | NULL | See the data_format parameter. |
| user_aa_composition | C2 H3 N1 O1 | Elemental formula of the form Cx Hy Oz etc where x, y and z are integers. Elements defined in params/elements.txt can be used. |
| user_aa_2_composition | "" | Elemental formula of the form Cx Hy Oz etc where x, y and z are integers. Elements defined in params/elements.txt can be used. |
| user_aa_3_composition | "" | Elemental formula of the form Cx Hy Oz etc where x, y and z are integers. Elements defined in params/elements.txt can be used. |
| user_aa_4_composition | "" | Elemental formula of the form Cx Hy Oz etc where x, y and z are integers. Elements defined in params/elements.txt can be used. |
| user_aa_5_composition | "" | Elemental formula of the form Cx Hy Oz etc where x, y and z are integers. Elements defined in params/elements.txt can be used. |
| user_aa_6_composition | "" | Elemental formula of the form Cx Hy Oz etc where x, y and z are integers. Elements defined in params/elements.txt can be used. |
| user_aa_7_composition | "" | Elemental formula of the form Cx Hy Oz etc where x, y and z are integers. Elements defined in params/elements.txt can be used. |
| user_aa_8_composition | "" | Elemental formula of the form Cx Hy Oz etc where x, y and z are integers. Elements defined in params/elements.txt can be used. |
| user_aa_9_composition | "" | Elemental formula of the form Cx Hy Oz etc where x, y and z are integers. Elements defined in params/elements.txt can be used. |
| dna_reading_frame | 1 | 1, 2, 3, 4, 5, 6 |
| open_reading_frame | 1 | non-zero positive integer |
| user_protein_sequence | "" | protein as a text string |
| hide_protein_sequence | 0 | 0, 1 |
| access_method | Index Number | Index Number Accession Number |
| entry_data | "" | See manual |
| index_num | 80707 | integer |
| accession_num | L39370 | text |
| MS-Product | ||
|---|---|---|
| name | Default Value | Valid Values |
| search_name | "" | text which can be part of a filename. |
| report_title | "" | needs to be set to msproduct |
| output_type | HTML | HTML, XML, Tab Delimited Text or mgf |
| script | "" | text |
| script_type | "" | text |
| instrument_name | "" | valid text strings from params/instrument.txt |
| results_to_file | 0 | 0, 1 |
| output_dir | "results_" + value of search_name parameter ie: results_msproduct | text |
| output_filename | "" | text |
| s | 1 | 0, 1 |
| sequence | SAMPLER | A,C,D,E,F,G,H,I,K,L,M,N,P,Q,R,S,T,V,W,Y m - Oxidized Methionine h - Homoserine lactone U - Selenocysteine s,t,y - Phosphorylated S,T,Y d, e, f, g, h, i, j, k, l - user specified amino acids Modified amino acids may be entered using PSI notation - eg. M(Oxidation), S(Phospho) An amino acid can also be followed by an exact mass - eg. P(-27.9949) or N(0.9840) |
| nterm | "" | N-terminus defined in params/usermod.txt, an exact mass or "" |
| cterm | "" | C-terminus defined in params/usermod.txt, an exact mass or "" |
| nloss | "" | Neutral loss defined in params/usermod.txt, an exact mass or "" |
| mods | "" | If there are multiple peptides it is possible either to specify each peptide individually (say a set of modification permutations) or to specify a value for the mods variable that describes the modifications and any ambiguity. The mods variable should be a combination of the const mod and variable mod columns in the Search Compare output. N-term, C-term and Neutral-loss are specified here. |
| s2 | 1 | 0, 1 |
| sequence2 | "" | see instructions for sequence above |
| nterm2 | "" | see instructions for n_term above |
| cterm2 | "" | see instructions for c_term above |
| nloss2 | "" | see instructions for n_loss above |
| s3 | 1 | 0, 1 |
| sequence3 | "" | see instructions for sequence above |
| nterm3 | "" | see instructions for n_term above |
| cterm3 | "" | see instructions for c_term above |
| nloss3 | "" | see instructions for n_loss above |
| s4 | 1 | 0, 1 |
| sequence4 | "" | see instructions for sequence above |
| nterm4 | "" | see instructions for n_term above |
| cterm4 | "" | see instructions for c_term above |
| nloss4 | "" | see instructions for n_loss above |
| s5 | 1 | 0, 1 |
| sequence5 | "" | see instructions for sequence above |
| nterm5 | "" | see instructions for n_term above |
| cterm5 | "" | see instructions for c_term above |
| nloss5 | "" | see instructions for n_loss above |
| s6 | 1 | 0, 1 |
| sequence6 | "" | see instructions for sequence above |
| nterm6 | "" | see instructions for n_term above |
| cterm6 | "" | see instructions for c_term above |
| nloss6 | "" | see instructions for n_loss above |
| alternative | 0 | 0 or 1 |
| discriminating | 0 | 0 or 1 |
| parent_mass_convert | monoisotopic | monoisotopic average Par(mi)Frag(av) Par(av)Frag(mi) |
| data_source | Data Paste Area | List of Files, Upload Data From File, Data Paste Area |
| upload_data | "" | file name |
| data_format | "" | M/Z Charge, M/Z Intensity Charge |
| data | NULL | See the data_format parameter.
For MS-Tag the first line contains the m/z and optionally the charge of the parent ion. |
| it | None Defined | a,a-H2O,a-NH3,a-H3PO4, b,b-H2O,b-NH3,b+H2O, b-H3PO4,b-SOCH4, y,y-H2O,y-NH3,y-H3PO4,y-SOCH4, MH+,B,c-1,c,c+1,c+2,x,Y,z,z+1,z+2,z+3,n,h,P,S,I,N,C |
| max_losses | 1 | 1,2,3 or All |
| max_internal_len | 200 | integer |
| msms_parent_mass_tolerance | 0.5 | double |
| msms_parent_mass_tolerance_units | Da | Da, %, ppm, mmu |
| fragment_masses_tolerance | 1.0 | double |
| fragment_masses_tolerance_units | Da | Da, %, ppm, mmu |
| user_aa_composition | C2 H3 N1 O1 | Elemental formula of the form Cx Hy Oz etc where x, y and z are integers. Elements defined in params/elements.txt can be used. |
| user_aa_2_composition | "" | Elemental formula of the form Cx Hy Oz etc where x, y and z are integers. Elements defined in params/elements.txt can be used. |
| user_aa_3_composition | "" | Elemental formula of the form Cx Hy Oz etc where x, y and z are integers. Elements defined in params/elements.txt can be used. |
| user_aa_4_composition | "" | Elemental formula of the form Cx Hy Oz etc where x, y and z are integers. Elements defined in params/elements.txt can be used. |
| user_aa_5_composition | "" | Elemental formula of the form Cx Hy Oz etc where x, y and z are integers. Elements defined in params/elements.txt can be used. |
| user_aa_6_composition | "" | Elemental formula of the form Cx Hy Oz etc where x, y and z are integers. Elements defined in params/elements.txt can be used. |
| user_aa_7_composition | "" | Elemental formula of the form Cx Hy Oz etc where x, y and z are integers. Elements defined in params/elements.txt can be used. |
| user_aa_8_composition | "" | Elemental formula of the form Cx Hy Oz etc where x, y and z are integers. Elements defined in params/elements.txt can be used. |
| user_aa_9_composition | "" | Elemental formula of the form Cx Hy Oz etc where x, y and z are integers. Elements defined in params/elements.txt can be used. |
| use_instrument_ion_types | 0 | 0,1 |
| max_charge | 1 | positive integer or No Limit |
| count_pos_z | Count Basic AA | Count Basic AA, Ignore Basic AA |
| msms_max_peaks | "" | integer |
| multi_z_internal | 0 | 0,1 |
| calibrate | 0 | 0,1 |
| cal_tolerance | 0.0 | double |
| data_plotted | Centroid | Centroid, Raw and Centroid, Raw |
| msms_pk_filter | Max MSMS Pks | Max MSMS Pks, Max MSMS Pks / 100 Da or Unprocessed MSMS |
| display_graph | 0 | 0,1 |
| search_key | "" | string |
| fraction | -1 | integer |
| spot_number | "1" | string |
| run | 1 | integer |
| spectrum_number | 1 | integer |
| msms_info | "" | string |
| msprod_min_mz | "" | double or blank |
| msprod_max_mz | "" | double or blank |
| msprod_min_z | "" | int or blank |
| msprod_max_z | "" | int or blank |
| link_search_type | No Link | valid text strings defined in links.txt and links_comb.txt |
| glyco | 0 | 0,1 |
| all_glyco | 0 | 0,1 |
MS-Comp is run using the executable file cgi-bin/mssearch.cgi. The program can either be run from the command line or via a HTML form. The HTML form for running it is generated by the program cgi-bin/msform.cgi. The default form settings are stored in the file params/mscomp/default.xml. The relevant parameter files are info.txt, aa.txt, elements.txt, imm.txt, instrument.txt and usermod.txt. Possible output formats are HTML or XML. The HTML report makes use of the cascade stylesheet files html/pform.css and html/print.css and the image file html/images/bannerblack.gif. The HTML report contains URL links to the MS-Isotope program. If the output is written to a file rather than to the standard output then the file is stored in the directory results/mscomp. The mssearch.cgi and msform.cgi programs can optionally write log files that are written to the directory logs/yyyy_mm_dd.
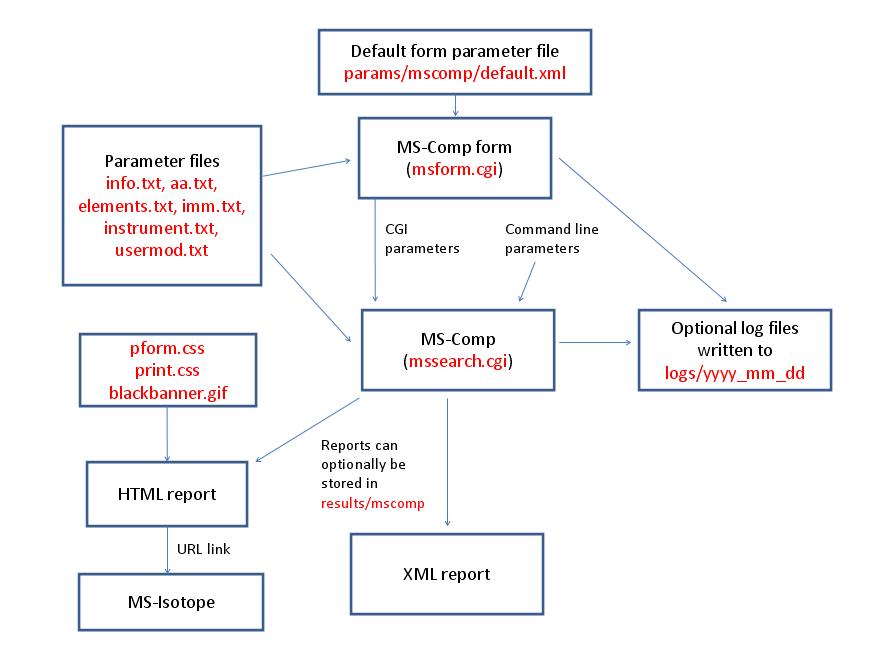
| MS-Comp | ||
|---|---|---|
| name | Default Value | Valid Values |
| search_name | "" | text which can be part of a filename. |
| report_title | "" | needs to be set to mscomp |
| output_type | HTML | HTML or XML |
| script | "" | text |
| script_type | "" | text |
| instrument_name | "" | valid text strings from params/instrument.txt |
| const_mod | None defined | valid text strings formed from the information in params/usermod.txt. |
| results_to_file | 0 | 0, 1 |
| output_dir | "results_" + value of search_name parameter ie: results_mscomp | text |
| output_filename | "" | text |
| max_reported_hits | 50 | integer |
| parent_mass_convert | monoisotopic | monoisotopic average Par(mi)Frag(av) Par(av)Frag(mi) |
| parent_mass_tolerance | 0.5 | double |
| tolerance_units | Da | Da, %, ppm, mmu |
| it | None Defined | a,a-H2O,a-NH3,a-H3PO4, b,b-H2O,b-NH3,b+H2O, b-H3PO4,b-SOCH4, y,y-H2O,y-NH3,y-H3PO4,y-SOCH4, MH+,c,B,n,h,P,S,I,N,C |
| comp_ion | None Defined | regular expression |
| parent_mass | 1000.0 | double |
| parent_charge | 1 | integer |
| composition_search | 0 | 0, 1 |
| aa_exclude | "" | text string containing the following characters A,C,D,E,F,G,H,I,K,L,M,N,P,Q,R,S,T,V,W,Y |
| aa_add | "" | d,e,f,g,h,i,j,k,l,m,s,t,y,z |
| user_aa_composition | C2 H3 N1 O1 | Elemental formula of the form Cx Hy Oz etc where x, y and z are integers. Elements defined in params/elements.txt can be used. |
| user_aa_2_composition | "" | Elemental formula of the form Cx Hy Oz etc where x, y and z are integers. Elements defined in params/elements.txt can be used. |
| user_aa_3_composition | "" | Elemental formula of the form Cx Hy Oz etc where x, y and z are integers. Elements defined in params/elements.txt can be used. |
| user_aa_4_composition | "" | Elemental formula of the form Cx Hy Oz etc where x, y and z are integers. Elements defined in params/elements.txt can be used. |
| user_aa_5_composition | "" | Elemental formula of the form Cx Hy Oz etc where x, y and z are integers. Elements defined in params/elements.txt can be used. |
| user_aa_6_composition | "" | Elemental formula of the form Cx Hy Oz etc where x, y and z are integers. Elements defined in params/elements.txt can be used. |
| user_aa_7_composition | "" | Elemental formula of the form Cx Hy Oz etc where x, y and z are integers. Elements defined in params/elements.txt can be used. |
| user_aa_8_composition | "" | Elemental formula of the form Cx Hy Oz etc where x, y and z are integers. Elements defined in params/elements.txt can be used. |
| user_aa_9_composition | "" | Elemental formula of the form Cx Hy Oz etc where x, y and z are integers. Elements defined in params/elements.txt can be used. |
| combination_type | Amino Acid | Amino Acid Peptide Elemental Elemental |
DB-Stat is run using the executable file cgi-bin/mssearch.cgi. The program can either be run from the command line or via a HTML form. The HTML form for running it is generated by the program cgi-bin/msform.cgi. The default form settings are stored in the file params/dbstat/default.xml. The relevant parameter files are info.txt, aa.txt, computer.txt, dbEST.spl.txt (dbEST database searches only), dbstat_hist.par.txt (HTML reports only), elements.txt, enzyme.txt, enzyme_comb.txt, taxonomy.txt and taxonomy_groups.txt (taxonomy searches only), and usermod.txt. The program also requires an indexed protein database produced by the FA-Index program. Possible output formats are HTML or XML. The HTML report makes use of either the Javascript file html/js/sGraph.js or the R script cgi-bin/peptideDensity.R dependent on the number of points in the graph. The data for the graph is temporarily written to a file in the directory temp/mmm_dd_yyyy. HTML reports also use the cascade stylesheet files html/pform.css and html/print.css and the image file html/images/bannerblack.gif. If the output is written to a file rather than to the standard output then the file is stored in the directory results/dbstat. The mssearch.cgi and msform.cgi programs can optionally write log files that are written to the directory logs/yyyy_mm_dd.
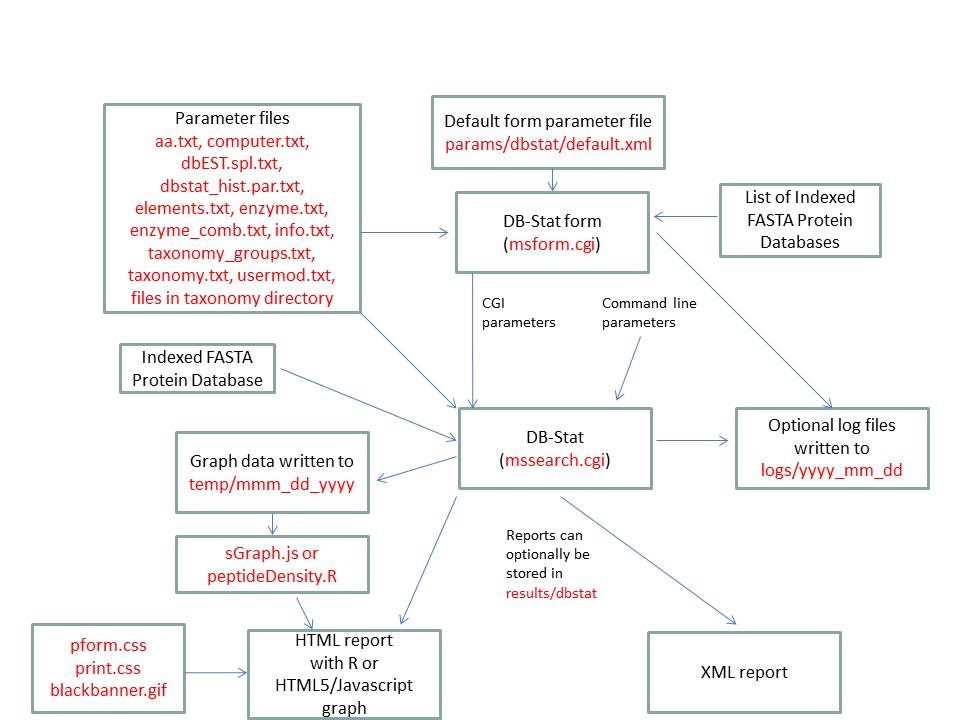
| DB-Stat | ||
|---|---|---|
| name | Default Value | Valid Values |
| search_name | "" | text which can be part of a filename. |
| report_title | "" | needs to be set to dbstat |
| output_type | HTML | HTML or XML |
| script | "" | text |
| script_type | "" | text |
| database | "" | valid prefixes: Genpept, gen, SwissProt, swp, Owl, owl, UniProt, Ludwignr, NCBInr, nr, dbEST, dbest, pdbEST, pdbest, IPI, ipi, DA, DN, PA, PN, pDA, pDN, Pdefault, Ddefault, pDdefault. User Protein is another possible selection. Multiple databases may be specified. |
| user_protein_sequence | "" | protein as a text string |
| results_from_file | 0 | 0, 1 |
| results_input_dir | results_" + value of input_program_name parameter eg: results_msfit | directory name |
| input_program_name | msfit | msfit, mstag, mspattern, mshomology, msseq |
| input_filename | "" | text |
| indicies | "" | list of database indicies (integers) |
| accession_numbers | "" | list of database accession numbers |
| species | All | valid text strings from params/taxonomy.txt, params/taxonomy_groups.txt or All |
| names | None Defined | list of text strings |
| accession_nums | None Defined | list of text strings |
| species_names | None Defined | list of text strings |
| species_remove | 0 | 0, 1 |
| add_accession_numbers | None Defined | list of text strings |
| prot_low_mass | 1000 | integer |
| prot_high_mass | 100000 | integer |
| full_mw_range | 0 | 0, 1 |
| low_pi | 3.0 | double |
| high_pi | 10.0 | double |
| full_pi_range | 0 | 0, 1 |
| enzyme | Trypsin | valid text strings from params/enzyme.txt or params/enzyme_comb.txt |
| missed_cleavages | 1 | integer |
| const_mod | None defined | valid text strings formed from the information in params/usermod.txt. |
| results_to_file | 0 | 0, 1 |
| output_dir | "results_" + value of search_name parameter ie: results_dbstat | text |
| output_filename | "" | text |
| dna_reading_frame | 1 | 1, 2, 3, 4, 5, 6 |
| show_aa_statistics | 0 | 0, 1 |
MS-Isotope is run using the executable file cgi-bin/mssearch.cgi. The program can either be run from the command line or via a HTML form. The HTML form for running it is generated by the program cgi-bin/msform.cgi. The default form settings are stored in the file params/msisotope/default.xml. The relevant parameter files are info.txt, aa.txt, elements.txt, sp_graph.par.txt and usermod.txt. Possible output formats are HTML, XML or Tab delimited text. The HTML report makes use of the Javascript file html/js/sGraph.js, the cascade stylesheet files html/pform.css and html/print.css and the image file html/images/bannerblack.gif. If the output is written to a file rather than to the standard output then the file is stored in the directory results/msisotope. The mssearch.cgi and msform.cgi programs can optionally write log files that are written to the directory logs/yyyy_mm_dd.
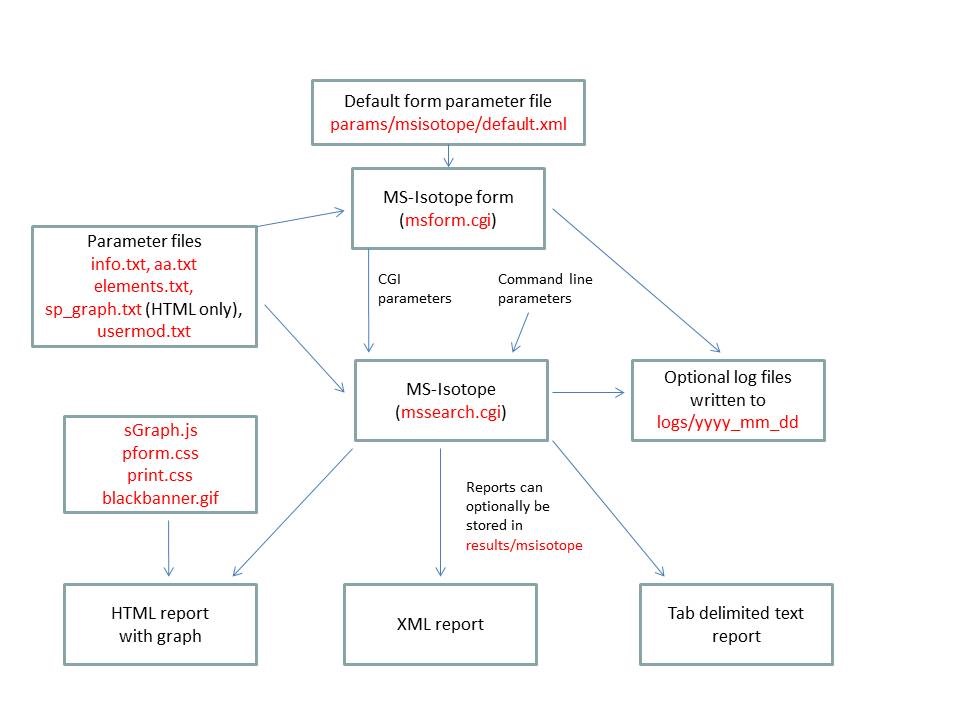
Note that the web version of MS-Isotope supports up to 6 isotope profiles using the parameters parent_charge, parent_intensity, distribution_type, nterm, sequence, cterm, elemental_composition and averagine_mass. For the second and subsequent profiles the parameter names are followed by an integer (eg. parent_charge2). For the command line version there is no limit on the number of profiles that can be used.
| MS-Isotope | ||
|---|---|---|
| name | Default Value | Valid Values |
| search_name | "" | text which can be part of a filename. |
| report_title | "" | needs to be set to msisotope |
| output_type | HTML | HTML, XML or Tab delimited text |
| script | "" | text |
| script_type | "" | text |
| results_to_file | 0 | 0, 1 |
| output_dir | "results_" + value of search_name parameter eg: results_msisotope | text |
| output_filename | "" | text |
| parent_charge | 1 | integer |
| parent_intensity | 1 | double |
| distribution_type | Peptide Sequence | Peptide Sequence, Elemental Composition |
| nterm | "" | N-termini defined in params/usermod.txt or "" |
| sequence | SAMPLER | A,C,D,E,F,G,H,I,K,L,M,N,P,Q,R,S,T,V,W,Y,d,e,f,g,h,i,j,k,l,m,s,t,y,z |
| cterm | "" | C-termini defined in params/usermod.txt or "" |
| elemental_composition | "" | Elemental formula of the form Cx Hy Oz etc where x, y and z are integers. Elements defined in params/elements.txt can be used. |
| averagine_mass | 1000.0 | double |
| parent_charge2 | 1 | integer |
| parent_intensity2 | 1 | double |
| distribution_type2 | Off | Peptide Sequence, Elemental Composition |
| nterm2 | "" | N-termini defined in params/usermod.txt or "" |
| sequence2 | "" | A,C,D,E,F,G,H,I,K,L,M,N,P,Q,R,S,T,V,W,Y,d,e,f,g,h,i,j,k,l,m,s,t,y,z |
| cterm2 | "" | C-termini defined in params/usermod.txt or "" |
| elemental_composition2 | "" | Elemental formula of the form Cx Hy Oz etc where x, y and z are integers. Elements defined in params/elements.txt can be used. |
| averagine_mass2 | 0.0 | double |
| parent_charge3 | 1 | integer |
| parent_intensity3 | 1 | double |
| distribution_type3 | Off | Peptide Sequence, Elemental Composition |
| nterm3 | "" | N-termini defined in params/usermod.txt or "" |
| sequence3 | "" | A,C,D,E,F,G,H,I,K,L,M,N,P,Q,R,S,T,V,W,Y,d,e,f,g,h,i,j,k,l,m,s,t,y,z |
| cterm3 | "" | C-termini defined in params/usermod.txt or "" |
| elemental_composition3 | "" | Elemental formula of the form Cx Hy Oz etc where x, y and z are integers. Elements defined in params/elements.txt can be used. |
| averagine_mass3 | 0.0 | double |
| parent_charge4 | 1 | integer |
| parent_intensity4 | 1 | double |
| distribution_type4 | Off | Peptide Sequence, Elemental Composition |
| nterm4 | "" | N-termini defined in params/usermod.txt or "" |
| sequence4 | "" | A,C,D,E,F,G,H,I,K,L,M,N,P,Q,R,S,T,V,W,Y,d,e,f,g,h,i,j,k,l,m,s,t,y,z |
| cterm4 | "" | C-termini defined in params/usermod.txt or "" |
| elemental_composition4 | "" | Elemental formula of the form Cx Hy Oz etc where x, y and z are integers. Elements defined in params/elements.txt can be used. |
| averagine_mass4 | 0.0 | double |
| parent_charge5 | 1 | integer |
| parent_intensity5 | 1 | double |
| distribution_type5 | Off | Peptide Sequence, Elemental Composition |
| nterm5 | "" | N-termini defined in params/usermod.txt or "" |
| sequence5 | "" | A,C,D,E,F,G,H,I,K,L,M,N,P,Q,R,S,T,V,W,Y,d,e,f,g,h,i,j,k,l,m,s,t,y,z |
| cterm5 | "" | C-termini defined in params/usermod.txt or "" |
| elemental_composition5 | "" | Elemental formula of the form Cx Hy Oz etc where x, y and z are integers. Elements defined in params/elements.txt can be used. |
| averagine_mass5 | 0.0 | double |
| parent_charge6 | 1 | integer |
| parent_intensity6 | 1 | double |
| distribution_type6 | Off | Peptide Sequence, Elemental Composition |
| nterm6 | "" | N-termini defined in params/usermod.txt or "" |
| sequence6 | "" | A,C,D,E,F,G,H,I,K,L,M,N,P,Q,R,S,T,V,W,Y,d,e,f,g,h,i,j,k,l,m,s,t,y,z |
| cterm6 | "" | C-termini defined in params/usermod.txt or "" |
| elemental_composition6 | "" | Elemental formula of the form Cx Hy Oz etc where x, y and z are integers. Elements defined in params/elements.txt can be used. |
| averagine_mass6 | 0.0 | double |
| user_aa_composition | C2 H3 N1 O1 | Elemental formula of the form Cx Hy Oz etc where x, y and z are integers. Elements defined in params/elements.txt can be used. |
| user_aa_2_composition | "" | Elemental formula of the form Cx Hy Oz etc where x, y and z are integers. Elements defined in params/elements.txt can be used. |
| user_aa_3_composition | "" | Elemental formula of the form Cx Hy Oz etc where x, y and z are integers. Elements defined in params/elements.txt can be used. |
| user_aa_4_composition | "" | Elemental formula of the form Cx Hy Oz etc where x, y and z are integers. Elements defined in params/elements.txt can be used. |
| user_aa_5_composition | "" | Elemental formula of the form Cx Hy Oz etc where x, y and z are integers. Elements defined in params/elements.txt can be used. |
| user_aa_6_composition | "" | Elemental formula of the form Cx Hy Oz etc where x, y and z are integers. Elements defined in params/elements.txt can be used. |
| user_aa_7_composition | "" | Elemental formula of the form Cx Hy Oz etc where x, y and z are integers. Elements defined in params/elements.txt can be used. |
| user_aa_8_composition | "" | Elemental formula of the form Cx Hy Oz etc where x, y and z are integers. Elements defined in params/elements.txt can be used. |
| user_aa_9_composition | "" | Elemental formula of the form Cx Hy Oz etc where x, y and z are integers. Elements defined in params/elements.txt can be used. |
| profile_type | Peptide Sequence | Stick, Gaussian, Lorentzian |
| resolution | 10000.0 | double |
| display_graph | 0 | 0, 1 |
| detailed_report | 0 | 0, 1 |
| percent_C13 | 100.0 | 0.0-100.0 |
| percent_N15 | 100.0 | 0.0-100.0 |
| percent_O18 | 100.0 | 0.0-100.0 |
MS-Filter is run using the executable file cgi-bin/mssearch.cgi. The program can only be run via a HTML form. The HTML form for running it is generated by the program cgi-bin/msform.cgi. The default form settings are stored in the file params/msfilter/default.xml. The relevant parameter files are info.txt, aa.txt, elements.txt, instrument.txt, fragmentation.txt and mgf.xml. The only possible output formats is HTML. The HTML report makes use of the cascade stylesheet files html/pform.css and html/print.css and the image file html/images/bannerblack.gif. The mssearch.cgi and msform.cgi programs can optionally write log files that are written to the directory logs/yyyy_mm_dd.
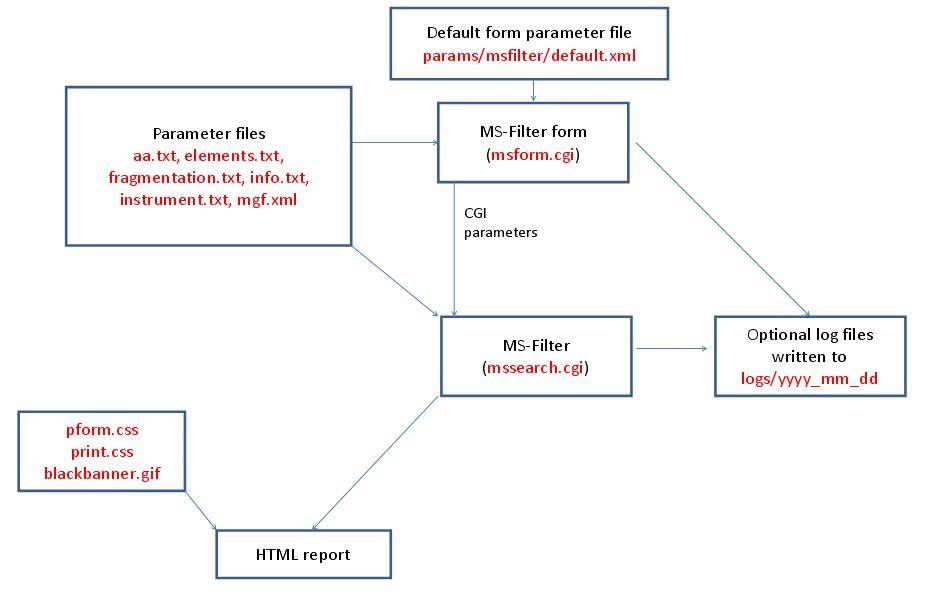
| MS-Filter | ||
|---|---|---|
| name | Default Value | Valid Values |
| report_title | "" | text |
| search_name | "" | text which can be part of a filename. |
| low_m_plus_h | 600.0 | double |
| high_m_plus_h | 4000.0 | double |
| full_m_plus_h_range | 0 | 0, 1 |
| charge_filter | None Defined | 1, 2, 3, 4, 5, 6, 7, 8, 9, 10 and above |
| all_charges | 0 | 0, 1 |
| loss_composition | "" | Elemental formula of the form Cx Hy Oz etc where x, y and z are integers. Elements defined in params/elements.txt can be used. |
| fragment_mz | 0.0 | double |
| instrument_name | "" | valid text strings from params/instrument.txt |
| parent_mass_convert | monoisotopic | monoisotopic average |
| fragment_masses_tolerance | 1.0 | double |
| fragment_masses_tolerance_units | Da | Da, %, ppm, mmu |
| msms_parent_mass_systematic_error | 0.0 | double |
| msms_parent_mass_tolerance_units | Da | Da, %, ppm, mmu |
| msms_pk_filter | Max MSMS Pks | Max MSMS Pks or Max MSMS Pks / 100 Da |
| msms_max_peaks | "" | integer |
| upload_temp_peak_list | "" | file name |
| msms_min_precursor_mass | 0 | 0, 1 |
| msms_precursor_exclusion | 0 | 0, 1 |
| keep_or_remove | Keep Spectra Matching Criteria | Keep Spectra Matching Criteria, Remove Spectra Matching Criteria or Create Both Sets of Spectra |
FA-Index is run using the executable file cgi-bin/faindex.cgi. The program can either be run from the command line or via a HTML form. The HTML form for running it is generated by the program cgi-bin/msform.cgi. The default form settings are built in. The relevant parameter files when creating indicies for a new database are info.txt, aa.txt, computer.txt, dbEST.spl.txt (dbEST databases only) and elements.txt. The program also requires a FASTA format protein database. The outputs are a HTML report and an indexed protein database for use by the Protein Prospector database search programs. HTML reports use the cascade stylesheet files html/pform.css and html/print.css and the image file html/images/bannerblack.gif. The faindex.cgi and msform.cgi programs can optionally write log files that are written to the directory logs/yyyy_mm_dd.
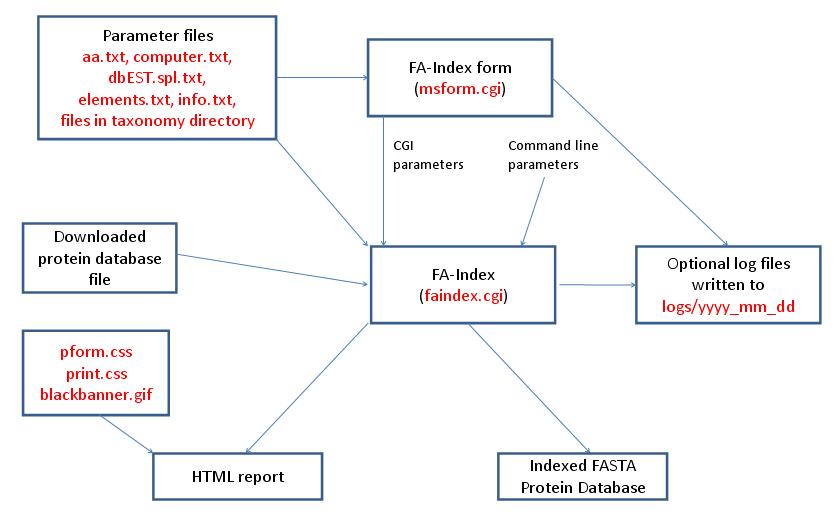
| FA-Index - Create Indicies for New Database | ||
|---|---|---|
| name | Default Value | Valid Values |
| database | "" | valid prefixes: Genpept, gen, SwissProt, swp, Owl, owl, UniProt, Ludwignr, NCBInr, nr, dbEST, dbest, pdbEST, pdbest, IPI, ipi, DA, DN, PA, PN, pDA, pDN, Pdefault, Ddefault, pDdefault. |
| dna_to_protein | 0 | 0, 1 |
| delete_dna_database | 0 | 0, 1 |
| random_database | 0 | 0, 1 |
| random_database | 0 | 0, 1 |
| use_seed | 0 | 0, 1 |
| seed | 0 | unsigned integer |
| reverse_database | 0 | 0, 1 |
| concat_database | 0 | 0, 1 |
| create_database_indicies | 0 | set this to 1 |
FA-Index is run using the executable file cgi-bin/faindex.cgi. The program can either be run from the command line or via a HTML form. The HTML form for running it is generated by the program cgi-bin/msform.cgi. The default form settings are built in. The relevant parameter files for a database summary report are info.txt, aa.txt, acclinks.txt, computer.txt, dbEST.spl.txt (dbEST databases only) and elements.txt and idxlinks.txt. The output is a HTML report. The HTML report uses the cascade stylesheet files html/pform.css and html/print.css and the image file html/images/bannerblack.gif. The faindex.cgi and msform.cgi programs can optionally write log files that are written to the directory logs/yyyy_mm_dd.
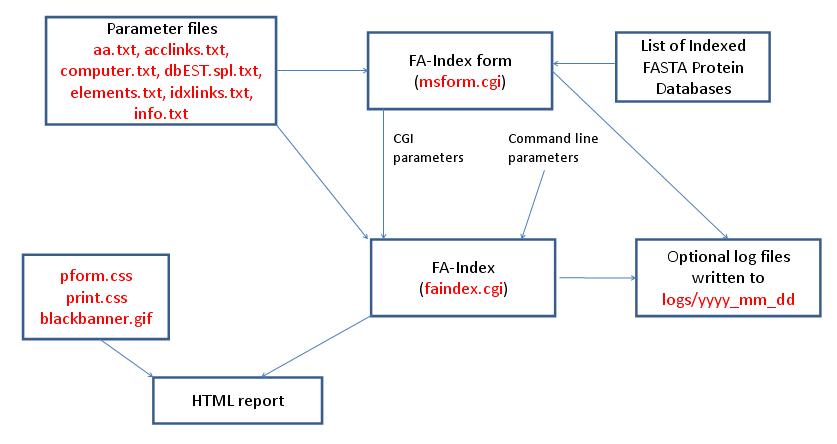
| FA-Index - Database Summary Report | ||
|---|---|---|
| name | Default Value | Valid Values |
| database | "" | valid prefixes: Genpept, gen, SwissProt, swp, Owl, owl, UniProt, Ludwignr, NCBInr, nr, dbEST, dbest, pdbEST, pdbest, IPI, ipi, DA, DN, PA, PN, pDA, pDN, Pdefault, Ddefault, pDdefault. |
| dna_reading_frame | 1 | 1, 2, 3, 4, 5, 6 |
| start_index_number | 1 | integer |
| end_index_number | 1 | integer |
| all_indicies | 0 | 0, 1 |
| hide_protein_sequence | 0 | 0, 1 |
FA-Index is run using the executable file cgi-bin/faindex.cgi. The program can either be run from the command line or via a HTML form. The HTML form for running it is generated by the program cgi-bin/msform.cgi. The default form settings are built in. The relevant parameter files when creating a subset database are info.txt, aa.txt, computer.txt, dbEST.spl.txt (dbEST databases only), elements.txt, taxonomy.txt and taxonomy_groups.txt (taxonomy searches only). The program also requires a FASTA formatted protein database that has already been indexed. The outputs are a HTML report and an indexed subset protein database for use by the Protein Prospector database search programs. The HTML report uses the cascade stylesheet files html/pform.css and html/print.css and the image file html/images/bannerblack.gif. The faindex.cgi and msform.cgi programs can optionally write log files that are written to the directory logs/yyyy_mm_dd.
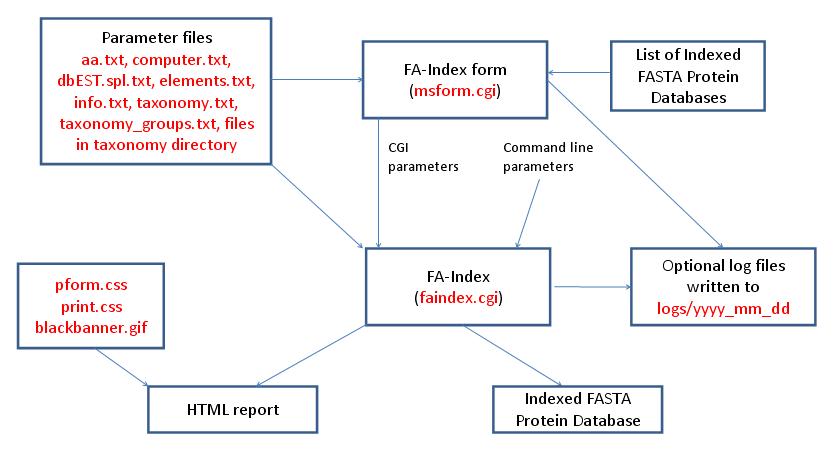
| FA-Index - Create Subset Database | ||
|---|---|---|
| name | Default Value | Valid Values |
| database | "" | valid prefixes: Genpept, gen, SwissProt, swp, Owl, owl, UniProt, Ludwignr, NCBInr, nr, dbEST, dbest, pdbEST, pdbest, IPI, ipi, DA, DN, PA, PN, pDA, pDN, Pdefault, Ddefault, pDdefault. |
| sub_database_id | sub | text |
| species | All | valid text strings from params/taxonomy.txt, params/taxonomy_groups.txt or All |
| species_remove | 0 | 0, 1 - Must be set to 1 for a DNA database |
| species_names | None Defined | list of text strings |
| prot_low_mass | 1000 | integer |
| prot_high_mass | 100000 | integer |
| full_mw_range | 0 | 0, 1 - Must be set to 1 for a DNA database |
| low_pi | 3.0 | double |
| high_pi | 10.0 | double |
| full_pi_range | 0 | 0, 1 |
| names | None Defined | list of text strings |
| accession_nums | None Defined | list of text strings |
| add_accession_numbers | None Defined | list of text strings |
| create_sub_database | 0 | set this to 1 |
| create_database_indicies | 0 | set this to 1 |
| FA-Index - Create Subset Database with Indices from Saved Hits | ||
|---|---|---|
| name | Default Value | Valid Values |
| database | "" | valid prefixes: Genpept, gen, SwissProt, swp, Owl, owl, UniProt, Ludwignr, NCBInr, nr, dbEST, dbest, pdbEST, pdbest, IPI, ipi, DA, DN, PA, PN, pDA, pDN, Pdefault, Ddefault, pDdefault. |
| results_from_file | 0 | 0, 1 |
| results_input_dir | results_" + value of input_program_name parameter eg: results_msfit | directory name |
| input_program_name | msfit | msfit, mstag, mspattern, mshomology, msseq |
| input_filename | "" | text |
| sub_database_id | .sub | text |
| create_database_indicies | 0 | set this to 1 |
| create_sub_database | 0 | set this to 1 |
| FA-Index - Create or Append to User Database | ||
|---|---|---|
| name | Default Value | Valid Values |
| database | "" | valid prefixes: Genpept, gen, SwissProt, swp, Owl, owl, UniProt, Ludwignr, NCBInr, nr, dbEST, dbest, pdbEST, pdbest, IPI, ipi, DA, DN, PA, PN, pDA, pDN, Pdefault, Ddefault, pDdefault. |
| name_field | "" | text |
| species | All | valid text strings from params/taxonomy.txt, params/taxonomy_groups.txt or All |
| accession_num | L39370 | text |
| user_protein_sequence | "" | protein as a text string |
| create_database_indicies | 0 | set this to 1 |
| create_user_database | 0 | set this to 1 |
 .
.